来源:https://baijiahao.baidu.com/s?id=1721157080537949425&wfr=spider&for=pc
药物发现的目的是寻找治疗疾病的具有特殊化学性质的新化合物。目前新药的发现和开发仍然是一个漫长而昂贵的过程,平均时间为10到15年。随着精准医学计划的开展,机器学习(ML)技术在药物发现领域得到了很大的重视,急剧降低了新药发现的成本和研究时间。ML是人工智能(AI)的一个分支,旨在开发和应用从原始数据中学习的计算机算法,以便后续执行特定任务。
今天给大家介绍的是Computational and Structural Biotechnology Journal杂志于2021年8月发表的综述《A review on Machine Learning approaches and trends in drug discovery》。第一作者为Paula Carracedo,西班牙拉科鲁尼亚大学。
机器学习方法
ML方法论在任何研究领域的应用都是横向的,意思是所有领域在实验设计中都有相同的步骤。具体地说,药物发现的ML方法可以分为以下5个步骤:数据收集、数学描述符的生成、搜索变量的最佳子集、模型训练和模型验证,如图1所示。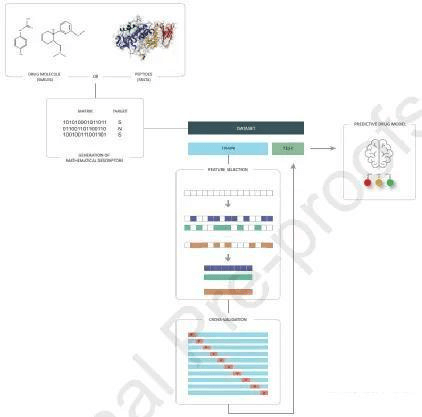
图1 药物发现中常用的机器学习方法
第一步是获取有特征的数据集,这些数据要求包括有助于吸收、特异性和低毒性的物理化学特性,以及易于在实验室生产和处理的特性。为了简化对这些化合物的处理和分析,分别使用SMILES和FASTA格式来表示小分子和肽的序列和结构。目前,存在许多存储用于药物发现领域数据的公共库,如DrugBank、PubChem、ChEMBL或ZINC。
新的测序技术在产生序列数据(DNA、RNA、蛋白质、小分子等)方面取得了很大进展。化合物序列是药物发现的起点,然后将序列转换成可由ML算法后续寻址的矩阵(图1 MATRIX),不同化合物的标签也很重要(图1 TARGET),因为在药物发现领域使用有监督学习模型较为常见。
数学描述符生成后,就得到了一组ML模型可以处理的数据。该数据集分为两个子集:较大的专用于训练模型(图1中蓝色),较小的子集用于测试模型(图1中绿色)。在训练集中,用正确和必要的信息搜索变量的最佳子集,尽可能减少无用或多余的变量,不同的技术如PCA、t-SNE、FS、自动编码器等。
一旦找到了变量的最佳子集,就对模型进行训练。首先,必须选择算法及其参数,常见的技术如交叉验证(CV),在实验执行过程中,原始数据集被再次划分为两个子集:训练集和验证集。图1展示了CV技术的10次运行,其中蓝色集合对应于训练集,红色集合对应于验证集。最终根据得到的最佳参数组合对每种模型的性能进行测量,最好的模型以最低成本实现最高性能价值。
最后,恢复从原始集合中提取的测试集(图1中绿色),并执行由CV过程产生的最佳模型的最终验证过程。如果验证结果具有统计学意义,则可以说已经创建了一个新的预测性药物模型。
机器学习用于分子表示
模型训练的关键一步依赖于分子的表示,这些表示需要捕捉分子的属性和结构特征,总的来说可以分为以下几类。
2.1 定量构效关系(QSAR)
在“分子结构决定其生物活性”和“结构相似的分子结构具有相似的生物活性”的前提下, QSAR可以通过数学系统,根据已知的化学结构和现有的实验研究,预测新化合物的理化和生物性质。QSAR在数值上将分子的化学结构与其生物活性联系起来。
要进行QSAR研究,需要三种类型的信息:一是具有共同作用机制的不同化合物的分子结构,二是每个配体的生物活性数据,最后是由一组数值变量描述的物理化学性质,由计算技术虚拟生成的分子结构获得。
2.2 分子描述符(MD)
MD在许多研究领域中起着关键作用,它们可以被定义为定量描述其物理化学信息的分子的数值表示。人们已经定义了数以千计的分子描述符,它们以不同的方式对分子进行编码。从应用角度可以分为两大类:一是实验测量类,如logP、摩尔折射率、偶极矩和极化率;二是包含物理化学性质的理论类,包括结构、拓扑(它的计算是通过图论完成的)、几何(从经验方案导出,编码分子参与不同类型相互作用的能力)、电子学和物理化学(分子在面对外部反应时的行为)。除此之外,也可以根据维度进行分类,如图2所示。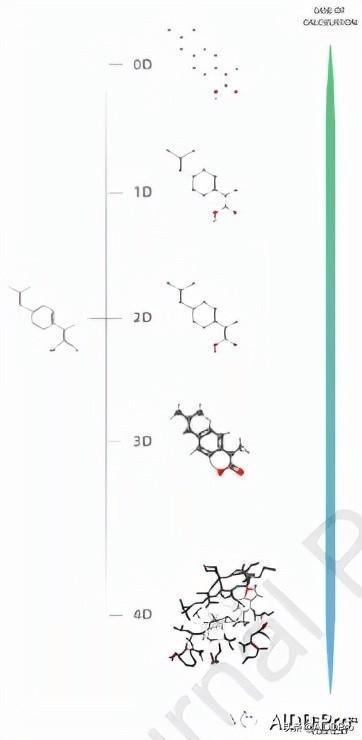
图2 由不同的分子描述符根据其维度编码的信息表示
2.3 分子指纹(FP)
FP允许快速而简单地通过固定长度的位链或矢量表示分子结构。这种形式的分子编码对于存储、处理和比较寄宿在包含分子信息的字符串中的数据是非常有效的。然而,来自化学结构的指纹忽略了生物背景,从而在分子结构和生物活性之间留下了鸿沟,因此前者的微小变化就可以产生生物活性的实质性差异。
FP种类繁多,最常用的包括扩展连通性指纹(ExtFP)、MACCS、PubChem指纹、原子对(包括APFP和GraphFP)、CDK、EstateFP和Klekota-Roth等。图3显示了几种方法的使用情况,可以看出,ExtFP指纹在绝对值上是使用最多的,然后是MACCS。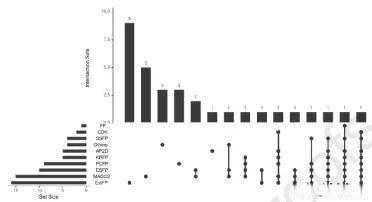
图3 常见FP的识别数量
2.4 基于图的机器学习算法
绝大多数分子描述符是以数字向量计算和编码的,生成高维矩阵用于经典ML算法,如随机森林、SVM、ANN、NB等。这些算法被设计为处理以矩阵或向量构成的数据,但不能使用以数学图形表示的分子的总信息。分子网络是化合物结构式的图论表示,每个分子用图表示,最近,能够预测特定功能的化学信息学模型的建立是基于从这些分子图中提取的信息,使用的算法是人工神经网络。
与从向量或数值矩阵中提取信息的全连接神经网络(FNN)或卷积神经网络(CNN)等更传统的拓扑结构不同,图形神经网络(GNN)能够从数学图中提取结构信息,最近开始在化学科学中引起极大的关注,如At Work[2]提出了一种基于深度度量学习的分子表示法,它可以自动为给定的数据集生成最优的表示法。通过这种方式,他们试图解决分子的性质对其分子结构的改变所产生的修饰。此外,这些算法还可以与其他算法相结合,有研究人员[3]基于GCN和强化学习进行新药设计,他们将产生具有所需相互作用性质的新分子作为一个多目标优化问题来处理。
这些技术的缺点是,大多数情况下,它们被视为黑盒,对结果的可解释性很低。目前有研究组正在开发新的用于分子表示的图形化神经网络体系结构,使用图形化的注意机制来从相关的药物发现数据集中学习,所获得结果的可解释性将大有提升。
机器学习用于生物学评估
药物可以被定义为一种与生物体中的一个功能实体相互作用的分子,这个实体称为治疗靶点或分子靶点。已知的药物作用于已知的靶点,发现可以改变疾病进程或提高现有治疗效果的新药物是化学和生物学领域研究的主要目标之一。
此前的统计数据表明,新药的发现和开发是一个非常复杂和昂贵的过程,完全是用实验方法进行的。过去几十年的技术进步促进了“Silico”的诞生,这个术语现在在生物实验室中很常见,指的是一种不是直接在活体上进行的实验(即活体实验),也不是在试管或有机体外的其他人工环境中进行的(即体外实验),而是通过计算机模拟生物过程进行的。现代生物学的复杂性使这些计算工具成为生物实验必不可少的工具,因为它们允许理论模型以极高的精度进行编码,并能够处理大量信息,从而促进和加快了新药开发的进程,主要应用于以下几个方面。
3.1 给药、分配、代谢、排出和毒性(ADMET)
药物相似度的概念是通过对已有或候选化合物的理化性质和结构特征的分析而建立的,已被广泛用于从ADMET等方面筛选出性能不佳的化合物。如拜耳制药公司已经在电子计算机上实现了一个ADME平台,目的是在药物发现的早期阶段为各种有用的药代动力学和物理化学特性建立模型。
3.2 抗菌剂&抗生素
近几十年来,几乎没有新的抗生素出现,而这种发现的速度跟不上日益增长的耐药性。然而,现有的大量数据促进了机器学习技术在药物发现中的应用(例如,建立回归、分类模型和化合物的虚拟分类或选择)。比如在抗击新冠中,冠状病毒的3C样蛋白酶(3CLpro)是一种潜在的抗SARS药物,已经开发了QSAR模型来寻找作为SARS-CoV-3CLpro酶抑制剂的化合物,并研究其抑制3CLpro的分子结构特征。
3.3 靶向药物相互作用
在开发一种疾病的治疗方法时,确定要达到的目标是最重要的,也是最容易出错的。化合物-蛋白质相互作用(CPI)分析已成为新药发现的重要前提,Multi-channel PINN等工具的开发在预测CPIs方面的进展为药物开发做出了巨大贡献。另一方面,鉴定目标蛋白的活性是药物发现的另一个早期步骤,eFindSite作为一种免费独立软件,使用有监督的机器学习来预测给定蛋白质的药理能力。此外,准确识别膜配体-蛋白结合位点将大大促进药物发现,MPLsPred工具利用多核方法对相互作用的预测使识别可能的药物-靶相互作用对成为可能。
3.4 抗癌
机器学习在癌症治疗领域也有重大贡献,激酶抑制剂在癌症药物发现领域拥有重要地位,工具诸如Kinformation基于机器学习方法,自动对激酶结构进行分类以改善蛋白激酶模型。另一方面,有研究人员提出新的计算方法,用于识别和表征肿瘤细胞间的通讯系统(如结缔组织)及其相关信号。此外,新的药物组合可以改善个性化癌症治疗,使用关于癌细胞系的各种类型的基因组信息、药物靶点和药理学信息,可以通过回归分类方法来预测药物组合的协同作用。
3.5 神经学疾病
药物神经毒性的计算模型在药物开发的早期被用于检测高风险化合物和选择更安全的候选药物。许多中枢神经系统疾病都需要多种策略来解决神经保护、修复和细胞再生问题。在神经退化过程和神经保护治疗中积累的知识,通过机器学习等计算技术来识别可以作为潜在的神经保护剂的药物组合。
3.6 药物重定位
药物重定位指的是开发一种用于治疗非目前适应症的化合物的过程,可以利用机器学习算法来学习现有药物相关生物数据中的模式,并将它们与要治疗的特定疾病联系起来。
4 ML在药物设计中的应用趋势
朴素贝叶斯模型已在药物发现中用于预测可能的药物靶点,已有团队[4]开发此类模型预测配体-靶相互作用,数据来自从KEGG和DrugBank获得的5125个已知与四种不同亚型蛋白质(酶、离子通道、GPCRs和核受体)的相互作用以及来自Stich的随机相互作用,准确率为95%。支持向量机(SVM)是生物信息学中应用最广泛的模型之一,因为它能够处理复杂的、非线性的、高维的和有噪声的问题,如通过从25个CHEMBL数据集中计算不同的分子描述符和化学指数来预测人肝微粒体的稳定性[5]。此外,无论要解决的问题类型是什么,尽管不可能将一个模型确定为适用于任何类型问题的最佳模型,但随机森林(RF)无疑是性能、速度和通用性方面最好的算法之一。而最近飞速发展的人工神经网络(ANN),基于深度学习技术,具有更灵活的架构,可以创建针对特定问题量身定制的NN架构,已成为药物发现的有用工具。
人工智能领域新算法设计的最新进展为解决不同学科的问题提供了机会。在化学信息学中,更具体地说,在药物发现中,这些模型的使用极大地造福了制药业(图4)。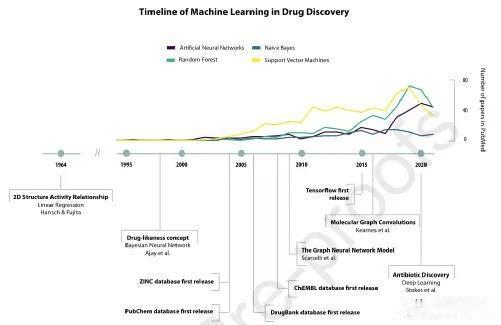
图4 药物发现领域机器学习重大事件时间表;折线图显示发表文献数量随时间的变化
值得强调的是,我们必须共同努力制定和使用标准化规则,这对于将学术成果转化为临床治疗具有重要意义。因此,机器学习技术的应用必须由不同领域的研究人员对实验进行全面的设计。总的来说,在精确医学和药物发现的背景下,ML技术提供的可能性和优势是巨大的。
参考文献:
[1] Paula Carracedo-Reboredo, Jose Liares-Blanco, A review on machine learning approaches and trends in drug discovery, Computational and Structural Biotechnology Journal, Volume 19,2021,Pages 4538-4558,ISSN 2001-0370
[2] G. S. Na, H. Chang, H. W. Kim, Machine-guided representation for accurate graph-based molecular machine learning, Physical Chemistry Chemical Physics 22 (33) (2020) 18526–18535.
[3] Y. Khemchandani, S. O’Hagan, S. Samanta, N. Swainston, T. J. Roberts, D. Bollegala, D. B. Kell, Deepgraphmolgen, a multiobjective, computational strategy for generating molecules with de- sirable properties: a graph convolution and reinforcement learning approach, Journal of Cheminformatics 12 (1) (2020) 1–17.
[4] Y. Wei, W. Li, T. Du, Z. Hong, J. Lin, Targeting hiv/hcv coinfection using a machine learning-based multiple quantitative structure-activity relationships (multiple qsar) method, International journalof molecular sciences 20 (14) (2019) 3572.
[5] I. Aliagas, A. Gobbi, T. Heffron, M.-L. Lee, D. F. Ortwine, M. Zak, S. C. Khojasteh, A probabilistic method to report predictions from a human liver microsomes stability qsar model: a practical tool for drugdiscovery,JournalofComputer-AidedMolecularDesign29(4) (2015) 327–338.
版 权 信 息
本文系AIDD Pro接受的外部投稿,文中所述观点仅代表作者本人观点,不代表AIDD Pro平台,如您发现发布内容有任何版权侵扰或者其他信息错误解读,请及时联系AIDD Pro (请添加微信号plgrace)进行删改处理。

若有收获,就点个赞吧
0 人点赞
