lncRNA鉴定
lncRNA特征
lncRNA是一类长度大于200个核苷酸并且不能翻译成蛋白质的转录本。根据lncRNA与蛋白编码基因在基因组上的位置关系,可以讲lncRNA分为五类:
- 反义lncRNA(antisense lncRNA,与mRNA所在位置相同,但是为反向互补关系)
- 增强子lncRNA(enhancer lncRNA,在mRNA的增强子区域)
- 基因间区lncRNA(intergenic lncRNA,在两个mRNA之间的基因间区)
- 双向lncRNA(bidirectional lncRNA,启动子区域与mRNA相同,但是为反向转录)
- 内含子lncRNA(intronic lncRNA,在mRNA的内含子区域)
目前研究发现,lncRNA具有调节基因表达、招募miRNA等功能,在生物发育过程中和重要形状/疾病中起重要的调控作用。与编码蛋白质的mRNA相比,lncRNA具有以下特点:
- 具有组织特异性
- 表达量低
- 不能编码蛋白质
- 物种之间保守性相对较差
- 转录本数量远多于mRNA等
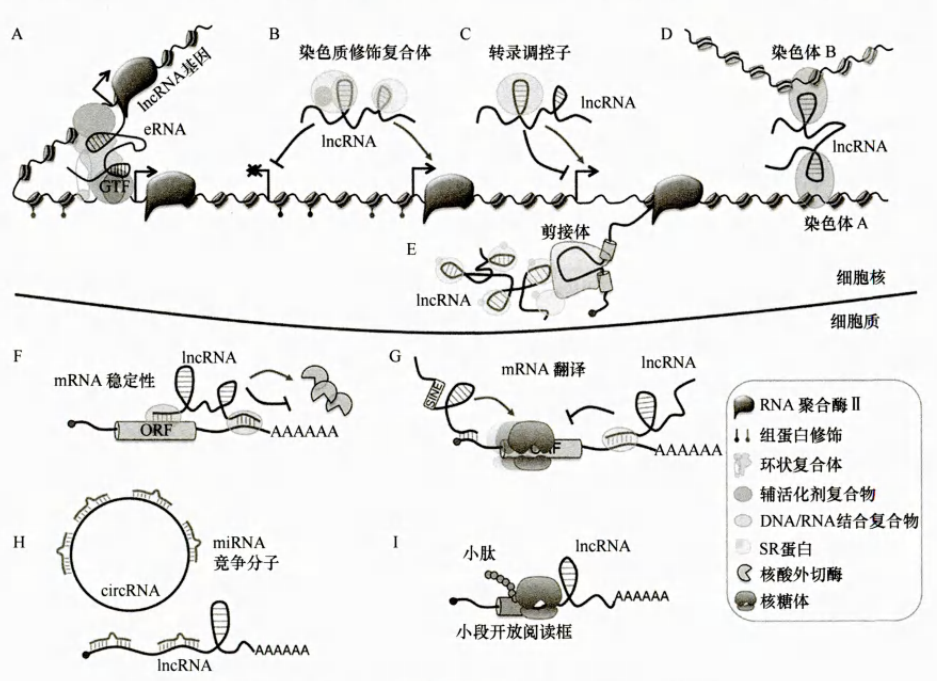
lncRNA的已知功能
A. lncRNA作为增强子来调节mRNA的转录;
B. lncRNA招募染色质修饰复合体来调节转录;
C. lncRNA通过调节转录因子活性来调节转录;
D. lncRNA通过改变染色体的空间结构来调节基因的表达;
E. lncRNA通过影响mRNA前体的剪接来影响基因的表达;
F. lncRNA通过调节mRNA的稳定性来调节mRNA的表达;
G. lncRNA通过调节mRNA的翻译来调节mRNA的表达;
H. lncRNA通过竞争性结合miRNA来调节mRNA的表达;
I. 一部分含有开放阅读框的lncRNA可以被翻译形成小肽。
lncRNA的鉴定方法
- 首先,需将转录组数据比对到基因组上
- 再利用Cufflink等转录组拼接软件可以得到新的转录本,用于之后lncRNA的甄别。
鉴定lncRNA最大的难点是确定转录组的非编码性,主要通过排除编码蛋白质的转录本来实现。对于编码蛋白质的mRNA来说,其开放阅读框(ORF)长度一般大于300nt,也就是说编码的蛋白质链长度大于100个氨基酸。因此,若RNA序列的ORF小于300nt,其编码蛋白质的可能性会非常小,会被判定为ncRNA。因此,若RNA序列的ORF小于300nt,其编码蛋白质的可能性会非常小,会被判定为ncRNA。然而这种武断的判断方法会存在一些问题,例如,有些lncRNA实际上其假定ORF长度要大于300nt,因此在该标准下它们会被错误划分为mRNA。类似的,有些ORF长度小于此阈值的mRNA会被误判为lncRAN。因此,可先根据ORF保守性,采用比较基因组学的方法进行甄别。mRAN的ORF具有保守性,即可编码蛋白质的转录本序列与已经注释的蛋白质或蛋白质结构域有相似性。因此可以采用BLASTX、Pfam等方法,将拼接后得到的转录组序列放到蛋白质库进行搜索,根据比对得到的同源相似性得分来判别是否可能编码蛋白质。不过值得注意的是,有些mRNA进化而来的lncRNA也会表现出与蛋白质序列相似的同源相似性,从而被错误判断为mRNA。目前可以采用综合性方法进行翻译潜能甄别,如利用CPC2、CONC、incRNA等软件,它们可以通过比较肽链长度、氨基酸构成、蛋白质同源性、二级结构、蛋白质比对或表达等多种特征来建立分类模型。CPC2基于序列内禀性质而不依赖其他外部数据,从而实现了物种无关性,同时速度也较CPC1提升近1000倍。
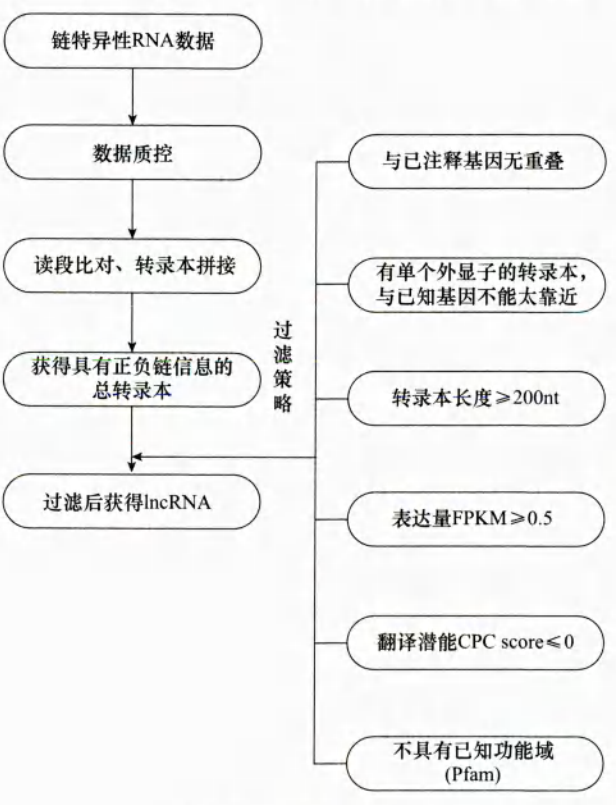
lncRNA鉴定流程
lncRNA功能预测
lncRNA与RNA分子互作预测
lncRNA作为miRNA的诱捕靶标
miRNA在动物和植物的生长发育中起着重要的调控作用。为了实现这些调控功能,miRNA先与AGO蛋白形成复合物,再通过碱基互补配对来绑定特定的信使RNA序列,导致信使RNA的翻译受阻或在特定位点剪接。
Franco-Zorrilla等在拟南芥中发现了一个由磷酸盐饥饿诱导不编码蛋白的基因IPS1。该基因能够与拟南芥中的miR399序列绑定在一起,但是在miR399的剪接位点形成了一个环状凸起结构。因此IPS1基因无法被切割,却能将miR399隔绝起来。而miR399真正的靶向基因是PHO2,该基因编码泛素合成酶,在维持细胞内蛋白质的产生和降解的平衡,以及维持细胞的稳态和正常方面起着重要作用。IPS基因的存在,使得miR399靶向PHO2基因的活性受到抑制,类似这种具有抑制miRNA功能的长非编码RNA定义为eTM(endogenous target mimics)。在动物中,常将这类lncRNA命名为miRNA海绵体或者miRNA诱饵,其与植物的区别主要还是在于miRNA与靶标序列结合的方式不同。
利用人工靶向基因模拟序列可以研究特定miRNA的功能,目前科学家已经对miRNA与其eTM的绑定规则有了一定研究,这为用生物信息学方法在植物体内大规模鉴定eTM建立了基础。基于已有研究,目前预测eTM主要采用如下方法:
- 首先,用FASTA3程序包中的搜索引擎获得与miRNA反向互补的cDNA序列,在搜索过程中允许互补位点有一个较大的凸起。
- 其次,对获得的序列进一步筛选,遵循如下规则:
在与相应miRNA互补配对的中间区域必须存在一个3~5个核苷酸的凸起;
除了中间的凸起区域,所有的错配数要小于4,且不允许产生连续两个错配;
除中间区域外,其他地方不允许产生凸起。
与miRNA预测方法的发展过程一样,随着实验验证结果的不断积累,eTM预测方法与在不断完善和改进。例如,新的研究建议遵循以下规则:
只允许在miRNA5’端序列上的第9~12个位点出现凸起;
eTM中的凸起部分由三个核苷酸组成;
在miRNA 5’端第2~8个位点要与eTM完全配对,但允许G/U错配;
除了中间凸起部分,其余错配数需不超过3,eTM的长度要大于200个核苷酸。
这些改进为今后全基因组上大规模鉴定eTM和验证其功能提供了更加完善的方法。
lncRNA与其他RNA分子互作预测
计算预测RNA-RNA的互作是基于分子间的相互作用能,它是通过对两个RNA分子内和分子间碱基配对时的结合能进行估计。
Busch等通过结合分子杂化自由能及互作时所需自由能,开发了IntaRNA算法。
Wright等(2013)在IntaRNA的基础上开发了CopraRNA,其主要改进是结合了比较基因组学的方法。
Terai等(2016)开发了一个计算流程,该流程结合了一系列软件用于预测人类中lncRNA-RNA的互作关系。不过在植物中,像这类预测方法和软件还有待深入研究。
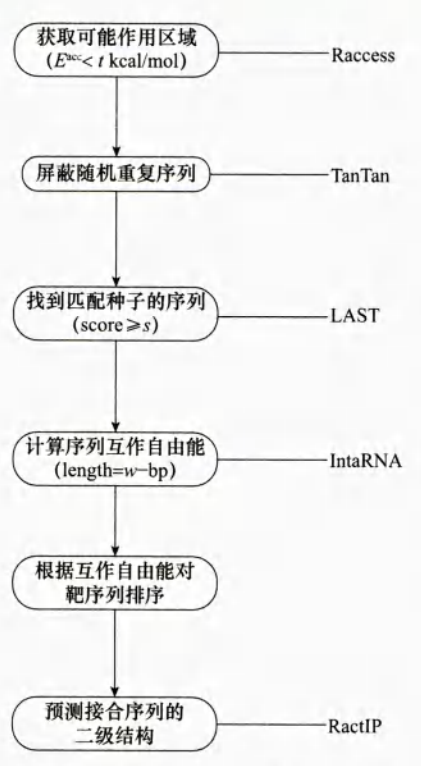
预测lncRNA与RNA互作的综合算法流程
lncRNA与蛋白质分子互作预测
已经有多个算法可以用来预测RNA与蛋白质互作关系。在这些算法中,机器学习方法(如Fisher线性判别分析、支持向量模型及随机森林)可以用来判断RNA与蛋白质是否互作。根据输入数据的不同格式,这些方法可以划分为三类:
第一类是基于序列的方法:RPI-seq、catRAPID和lncPRO都是基于序列所开发的方法,它们只需将RNA及蛋白质序列作为输入数据。在三类方法中,catRAPID和lncPRO利用氨基酸和核苷酸的物理化学特征来预测蛋白质和RNA的二级结构,作为判别RNA与蛋白质是否互作的证据。
第二类是基于序列和结构的方法:RPI-seq在预测RNA与蛋白质互作时,不仅用到了它们的序列信息,还需要将RNA与蛋白质的三维结构作为输入数据。在RPI-seq方法里,通过三维结构可以得到蛋白质结构域及RNA的二级结构,这些可以作为判别RNA与蛋白质是否作用成对的特征。相对来说,该方法准确性更高,但难度更大,因为目前只有为数不多的lncRNA结构注释信息。
第三类是基于实验数据的方法:Pancaldi和Bahler(2011)开发了基于多种实验数据的预测方法,这些实验数据包括蛋白质定位、RNA半衰期、核糖体分析及帕尔斯分析。对于该方法来说,由于可应用的数据更有限,所以难度就更大了。
可见机器学习方法将会更适应以后的发展趋势。为了评估预测准确性,还需要实验或者其他方法交叉验证。从这方面来看,用生物信息学的方法预测lncRNA与蛋白质的互作关系还是一个挑战,值得深入思考。
lncRNA功能注释在线平台
北京大学高歌团队开发的国际上首个lncRNA在线注释平台AnnoLnc,其第一个版本于2016年上线,第二个版本AnnoLnc2于2020年5月上线,该版本同时包含人类和小鼠的lncRAN。AnnoLnc2的使用十分便捷,只需提交序列,选择相应的物种,便可以对该lncRNA的结构、表达、功能和进化方面进行注释,具体注释的内容包括10个模块,即
- lncRNA的基因组位置
- 序列二级结构
- lncRNA与蛋白质互作
- 共表达
- GO富集分析
- lncRNA在不同组织(或细胞)中的表达
- 亚细胞定位
- 转录调节
- miRNA绑定调节
- 遗传关联及进化分析
此外,AnnoLnc2还有本地版本,支持批量处理数据,并且可以自定义模块和自定义注释数据。目前还未见用于植物lncRNA功能注释的在线平台。
参考
《生物信息学 第二版》樊龙江
综述 | Long Noncoding RNAs in Plants(公众号介绍)
北大 Gao Lab 工具网址:http://gsds.cbi.pku.edu.cn/

若有收获,就点个赞吧
0 人点赞
