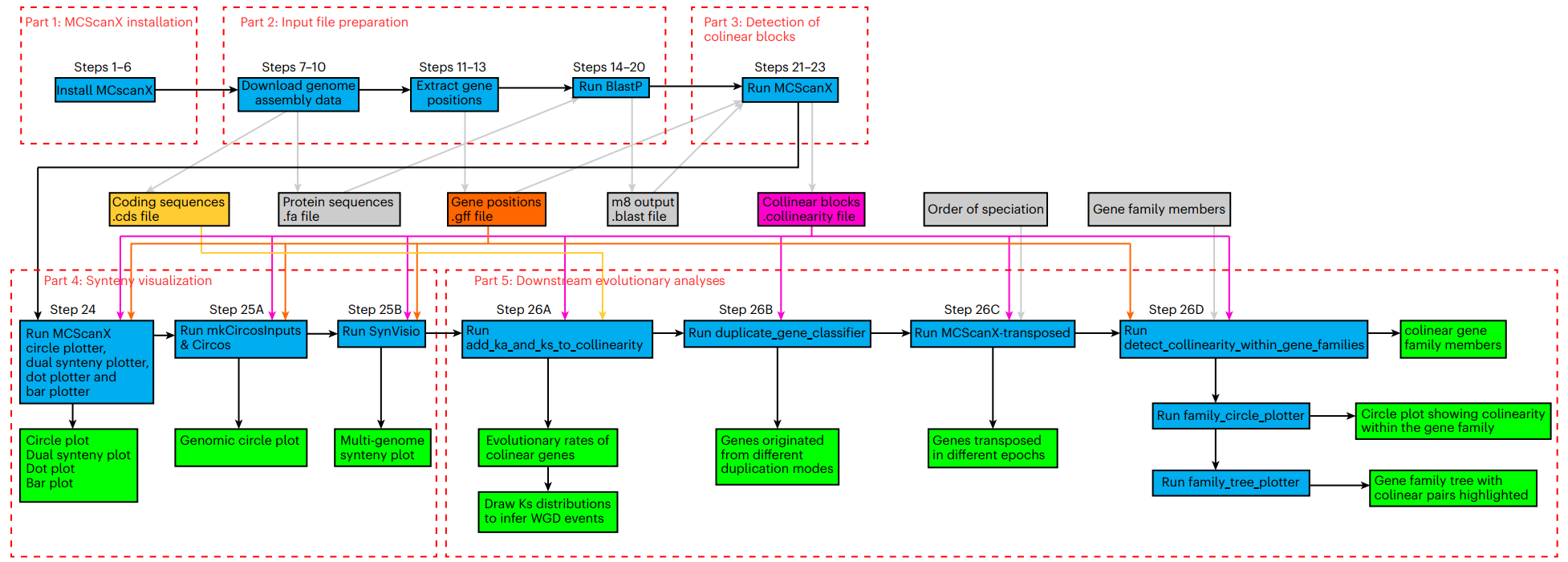
介绍
MCScanX

下载
# module load GCCcore/13.2.0# module spider Java/17.0.6git clone https://github.com/wyp1125/MCScanXcd MCScanxmakeexport PATH=$PATH:/proj/nobackup/hpc2nstor2024-021/shwzhao/bin/software/MCScanX
使用
MCScanX -h## [Usage] ./MCScanX prefix_fn [options]## -k MATCH_SCORE, final score=MATCH_SCORE+NUM_GAPS*GAP_PENALTY## (default: 50)## -g GAP_PENALTY, gap penalty (default: -1)## -s MATCH_SIZE, number of genes required to call a collinear block## (default: 5)## -e E_VALUE, alignment significance (default: 1e-05)## -m MAX_GAPS, maximum gaps allowed (default: 25)## -w OVERLAP_WINDOW, maximum distance (# of genes) to collapse BLAST matches (default: 5)## -a only builds the pairwise blocks (.collinearity file)## -b patterns of collinear blocks. 0:intra- and inter-species (default); 1:intra-species; 2:inter-species## -h print this help page
-k: MATCH_SCORE,匹配得分。[50]
指的是共线性区块内共线性基因对的得分。然而,两个连续的共线性基因对之间可能会出现间隙(即非共线性基因),这些间隙需要被惩罚。
-g: GAP_PENALTY,Gap 罚分。[-1]
对于一个共线性基因对,其 final_score 定义为 MATCH_SCORE + NUM_GAPS · GAP_PENALTY。对于一个报告的共线性区块,通过将其所有共线性基因对的 final scores 相加生成总得分。因此,为了提供更宽松的共线性基因比对,可以将 GAP_PENALTY 调整为比默认值(即 -1)更小的绝对值(例如 -0.5)。
-s: MATCH_SIZE,匹配大小。[5]
表示推断一个共线性区块所需的基因数量。增加 MATCH_SIZE 会减少检测到的共线性区块数量。对于小型基因组,可以将此选项调到比默认值更小的值(例如 4)。
-e: E-value,表示共线性区块基因比对的统计阈值。[1e-05]
增加此值会导致检测到更多的共线性区块,而减少此值会导致检测到更少的共线性区块。CUTOFF_score 定义为 MATCH_SIZE * MATCH_SCORE。如果一个共线性区块的得分未超过 CUTOFF_score,则不会被报告。
-m: MAX_GAPS,指定允许连接下一个共线性基因对的最大间隙。[25]
如果遇到的间隙超过 MAX_GAPS,则共线性区块将停止扩展。
-w: OVERLAP_WINDOW,。[5]-a:-b: 共线性块模式。[0]
0: 物种内和物种间;1: 物种内;2: 物种间。
-h: 显示帮助信息。
运行
数据下载
Arabidopsis thaliana 5 GCA_000001735.2 https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001735.4/ atArabidopsis suecica 13 GCA_019202805.1 https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_019202805.1/ asArabidopsis arenosa 8 GCA_905216605.1 https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_905216605.1/ aaThlaspi arvense 7 GCA_911865555.2 https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_911865555.2/ taBrassica oleracea var.oleracea 9 GCA_000695525.1 https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000695525.1/ boBrassica carinata 17 GCA_016771965.1 https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_016771965.1/ bc
# Arabidopsis_thalianawget -c https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/735/GCF_000001735.4_TAIR10.1/GCF_000001735.4_TAIR10.1_genomic.gff.gz -O Arabidopsis_thaliana.gff.gzwget -c https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/735/GCF_000001735.4_TAIR10.1/GCF_000001735.4_TAIR10.1_protein.faa.gz -O Arabidopsis_thaliana.pep.fa.gz# Arabidopsis_suecicawget -c https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/019/202/805/GCA_019202805.1_ASM1920280v1/GCA_019202805.1_ASM1920280v1_genomic.gff.gz -O Arabidopsis_suecica.gff.gzwget -c https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/019/202/805/GCA_019202805.1_ASM1920280v1/GCA_019202805.1_ASM1920280v1_protein.faa.gz -O Arabidopsis_suecica.pep.fa.gzgunzip *.gz
提取位置信息
blastp 比对
# module load GCC/12.3.0# module load OpenMPI/4.1.5# module load BLAST+/2.14.1cat Arabidopsis_thaliana.pep.fa Arabidopsis_suecica.pep.fa > all.pep.famakeblastdb -in all.pep.fa -dbtype prot -parse_seqids -out allblastp -query all.pep.fa -db Artha -outfmt 6 -out "all.blastp_all.tab" -num_threads 20 -evalue 1e-10
blastp \-db ncbiDB/species1 \-query ncbi/species2.fa \-evalue 1e-10 \-num_alignments 5 \-outfmt 6 \-out intermediateData/species1-2.blast
对于每一对基因组,调整 query 和 target 基因组,再进行第二次运行。
共线性块检测
800 //plot width and height (in pixels)at1,at2,at3,at4,at5,aa1,aa2,aa3,aa4,aa5 //chromosomes in the circle
java dual_synteny_plotter -g .gff -s .collinearity -c .ctl -o _synteny.pngjava dot_plotter -g.gff -s .collinearity -c .ctl -o _dot.pngjava circle_plotter -g .gff -s .collinearity -c .ctl -o _circle.pngjava bar_plotter -g .gff -s .collinearity -c .ctl -o _bar.png
perl add_Ka_and_Ks_to_collinearity_Yn00.pl \-i sp.collinearity \-d sp.cds \-o sp.KaKs
./duplicate_gene_classifier data/atperl MCScanX-transposed.pl -i dir -t target_species -c outgroup1,outgroup2,…,outgroupN -x 2 -o outputDirperl detect_colinearity_within_gene_families.pl -i gene.family -d .collinearity -o collinearity_within_gene_familiesjava family_circle_plotter -g .gff -s .collinearity -c family.ctl -f gene.family -o gene_family_circle_plot.pngjava family_tree_plotter -t family.tree -s .collinearity -d .tandem -x 400 -y 1100 -f 10 -o family_treePlot.png
参考
- https://github.com/wyp1125/MCScanX
- Detection of colinear blocks and synteny and evolutionary analyses based on utilization of MCScanX
- 基因组分析 | 基于 MCScanX 的共线区块检测和进化分析
conda create -n comparative_genomics
conda activate comparative_genomics

若有收获,就点个赞吧
0 人点赞
