染色质可及性测序技术的发展
什么是染色质可及性/开放性?真核生物中的核小体是染色质的基本结构单位。DNA 与组蛋白结合后形成核小体,核小体再进一步折叠压缩后最终形成染色质。DNA 的复制和转录都需要将致密染色质结构打开,从而允许转录因子和其他调控元件结合 DNA,这部分被打开的染色质,叫开放染色质区域(Open Chromatin Region),开放染色质区域允许调控因子结合的特性称为染色质可及性(Chromatin Accessibility)。因此与转录调控密切相关。
基于高通量测序技术的发展,检测染色质开放性的技术主要有以下五种:
- 脱氧核糖核酸酶 I 超敏感位点测序(DNase-seq)
- 甲醛辅助性调控元件分离测序(FAIRE-seq)
- 微球菌核酸酶敏感位点测序(MNase-seq)
- 核小体定位和甲基化测序(NOMe-seq)
- 染色质转座酶可及性测序(ATAC-seq)
事实上,按照获取染色质开放信息的方式,可以将上述五种技术分为3种:
- 酶切法(DNase-seq、MNase-seq、ATAC-seq)
- 超声断裂法(FAIRE-seq)
- 甲基化修饰(NOMe-seq)
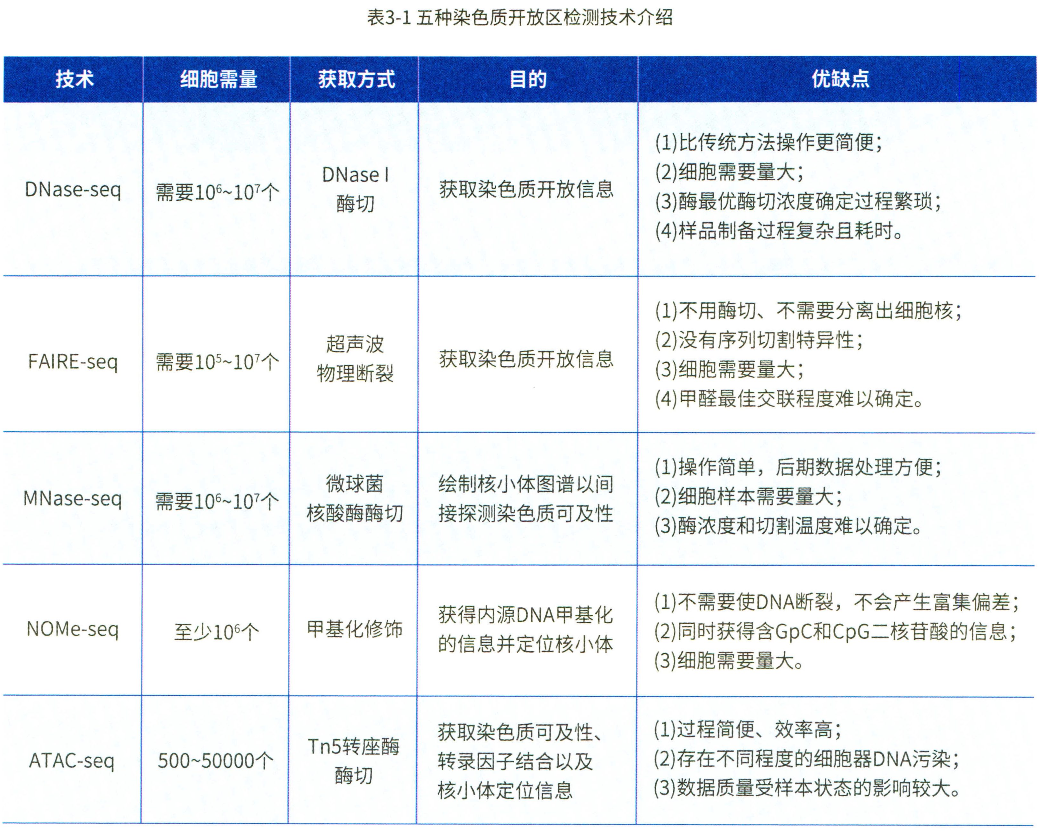
DNase-seq 技术
DNase I 超敏位点测序(DNase-seq)是研究开放染色质区域最经典的技术之一,DNase I 具有核酸内切酶活性,可通过控制切割效率获得适当长度的开放染色质片段。DNase I 被用来切割基因组上的 DNA 酶敏感部位,然后对消化的片段进行扩增,分析测序数据中的峰,以获得相对开放的染色质区域和蛋白质保护区域的信息,这些区域通常是转录因子结合的位点。但是它存在明显的技术弊端,如整个样品制备过程复杂且耗时、需要大量细胞、确定最优酶切浓度的过程也较为繁琐。
FAIRE-seq 技术
甲醛辅助分离调控元件技术(FAIRE-seq)是在全基因组范围内鉴定染色质可及性、检测与调节活性相关的 DNA 序列的方法。FAIRE-seq 技术检测开放染色质的方式与 DNase-seq 相似,但其操作过程比 DNase-seq 简便,它是使用超声波破碎经甲醛固定后的染色质,得到片段化的染色质,然后通过酚氯仿抽提,上层水相中即认为是潜在的开放染色质区,然后针对开放染色质区进行建库测序,获得开放区信息。该技术也同样存在细胞量需求大的问题,且超声打断DNA片段不易控制,最大的不足点在于甲醛最佳交联浓度和最佳交联时间的把握,因为过度交联或交联不充分都会影响最终的测序结果。
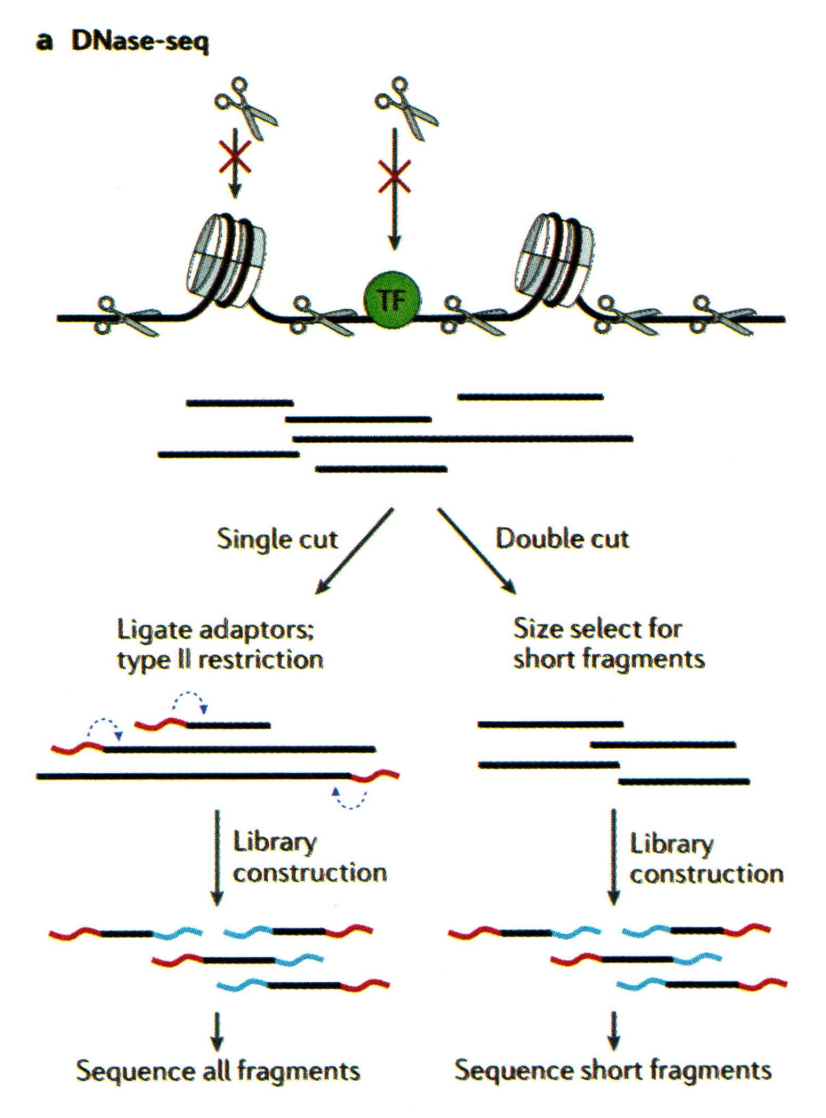
MNase-seq 技术
MNase-seq 是一种间接的检测开放染色质区域的技术,原理与 DNase-seq 类似,不过两种技术探测的区域是互补的。MNAase-seq 使用的是微球菌核酸酶(MNase),这是一种同时具有内切和外切酶活性的核酸酶,它能逐步切碎不被核小体或其他蛋白保护的 DNA,而被核小体或其他蛋白保护的 DNA 则不被酶切降解。将切割下来的 DNA 片段进行测序,与已知的全基因组序列进行比对,就可以知道没被切掉的是哪些位置,被切掉的位置在哪里,从而获得开放区的信息。因此该技术主要用于探究核小体的位置,开放区信号本身是缺失的(被 MNase 完全消化了)。但 MNase 偏好切割基因组中富含 AT 的区域,进而对核小体边界的精准确定产生影响,且该技术同样受到细胞量大、酶切浓度和酶切温度的限制。
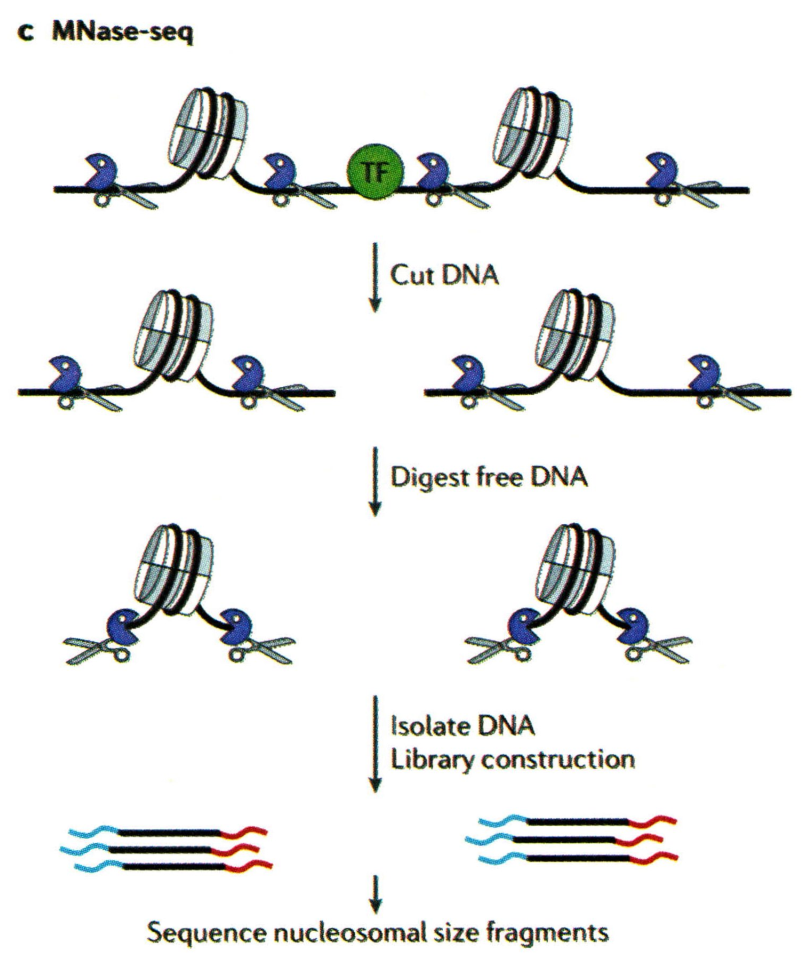
NOMe-seq 技术
NOMe-seq 于 2012 年 Kelly 等发明,是一种能在同一个 DNA 分子中观察到 DNA 甲基化和核小体定位的技术。NOMe-seq 方法基于用 GpC 甲基转移酶处理染色质,该酶不受核小体或其他与染色质紧密结合的蛋白质保护的 GpC 二核苷酸甲基化。随后,对 M.CviPI 甲基化染色质进行亚硫酸氢盐处理和全基因组测序,可以在全基因组范围内确定核小体耗尽区域。亚硫酸氢盐处理还可以区分 CpG 和 CmpG(大多数内源甲基化发生在 CpG 二核苷酸上),因此基因组的内源甲基化状态也可以在相同的测序反应中获得。由于 M.CiPI 处理是在 DNA 片段化之前进行的,因此 NOMe-seq 中对开放染色质的偏向较小,并可能存在较少的假阳性识别区域。
ATAC-seq 及其衍生技术
ATAC-seq 利用 Tn5 转座酶仅从 500~50000 个细胞中实现了对开放染色质、DNA 结合蛋白和核小体定位的表观基因组分析,文库制备过程简单有效,仅需两步即可完成:转座和 PCR。
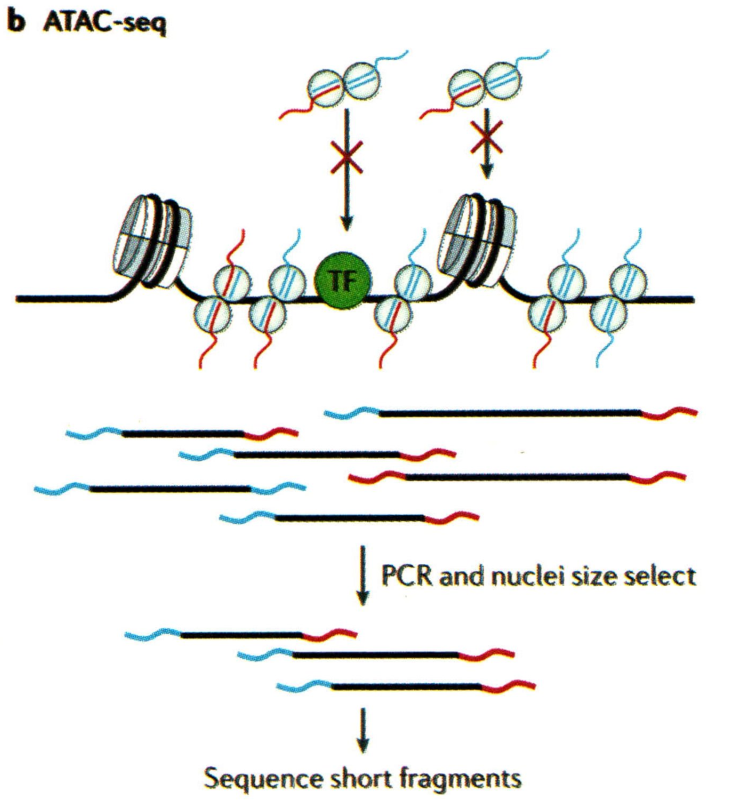
ATAC-seq 具有高效率和低细胞起适量的有点,但其适用性仍然有限制,为扩展应用而产生了许多 ATAC 衍生技术。
- Fast-ATAC
- Omni-ATAC
- miniATAC-seq
- ATAC-seq
ATAC-seq 实验原理
Tn5 转座酶的作用机制
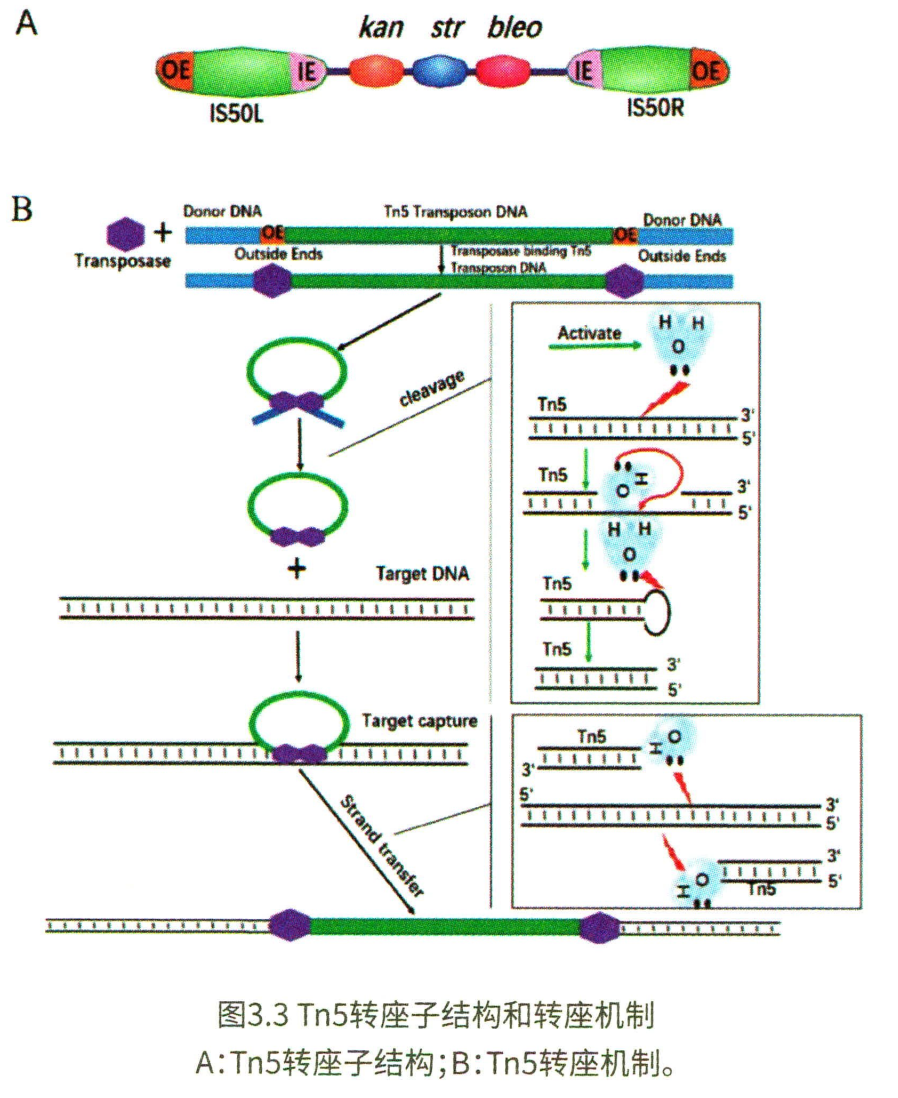
20世纪40年代,美国遗传学家 Barbara McClintock 在玉米中发现了第一个转座子。转座子是可以“跳跃”到基因组不同位置的遗传元件,故也叫跳跃基因或转座因子。根据转座子的结构特点和转座方式可将其分为 I 型和 II 型。
- I 型转座子,也称作 RNA 转座子(即反转录转座子),转座方式为“复制-粘贴”型,即在转座时以自身 DNA 为模板,在 RNA 聚合酶 II 的作用下,转录成一段 mRNA,然后以这段 mRNA 为模板反转录成 cDNA,也就是增加了自身一倍的拷贝数,最后在整合酶的作用下将这段增加的 cDNA 整合到基因组上新的位置。
- II 型转座子,也称作 DNA 转座子,直接以“剪切-粘贴”的方式剪切下自身的 DNA 序列插入到新的位置,不会增加拷贝数,其中 Helitron 转座子时近年来发现的一种新型 DNA 转座子,其结构特征具有典型的 5’-TC 以及 3’-CTRR(R位A或G)末端,并在 3’末端上有 10~12 个核苷酸的位置有一个 16~20bp 的颈环结构,是转座子的终止信号。Helitron 转座子转座后,通常插入 AT 丰富区域,Helitron 通过滚环(rolling circle)复制的方式进行转座扩增,并在扩增过程中经常捕获和携带基因片段,可导致基因拷贝数的变化,也会在一定程度上促进基因组的进化。
Tn5 转座子是一种细菌型转座子,属于 II 型转座子,最早是在大肠杆菌中发现的。它由编码卡那霉素、新霉素、链霉素 3 种抗生素的核心序列和位于侧翼的两个高度同源且倒置的 IS50(IS50L 和 IS50R)序列组成。其中,IS50 序列可编码参与转座的蛋白:转座酶(transposase,Tnp)和转座阻遏蛋白(transposase inhibitor,Inh)。但由于左侧末端的 IS50L 序列能够表达正常有活性的 Tnp 和 Inh。每个 IS50 序列具有两个 19bp 的倒置末端:外末端(outside end,OE)和内末端(inside end,IE)。两倒置末端有 7 个碱基不同,外末端是 Tnp 的结合位点。
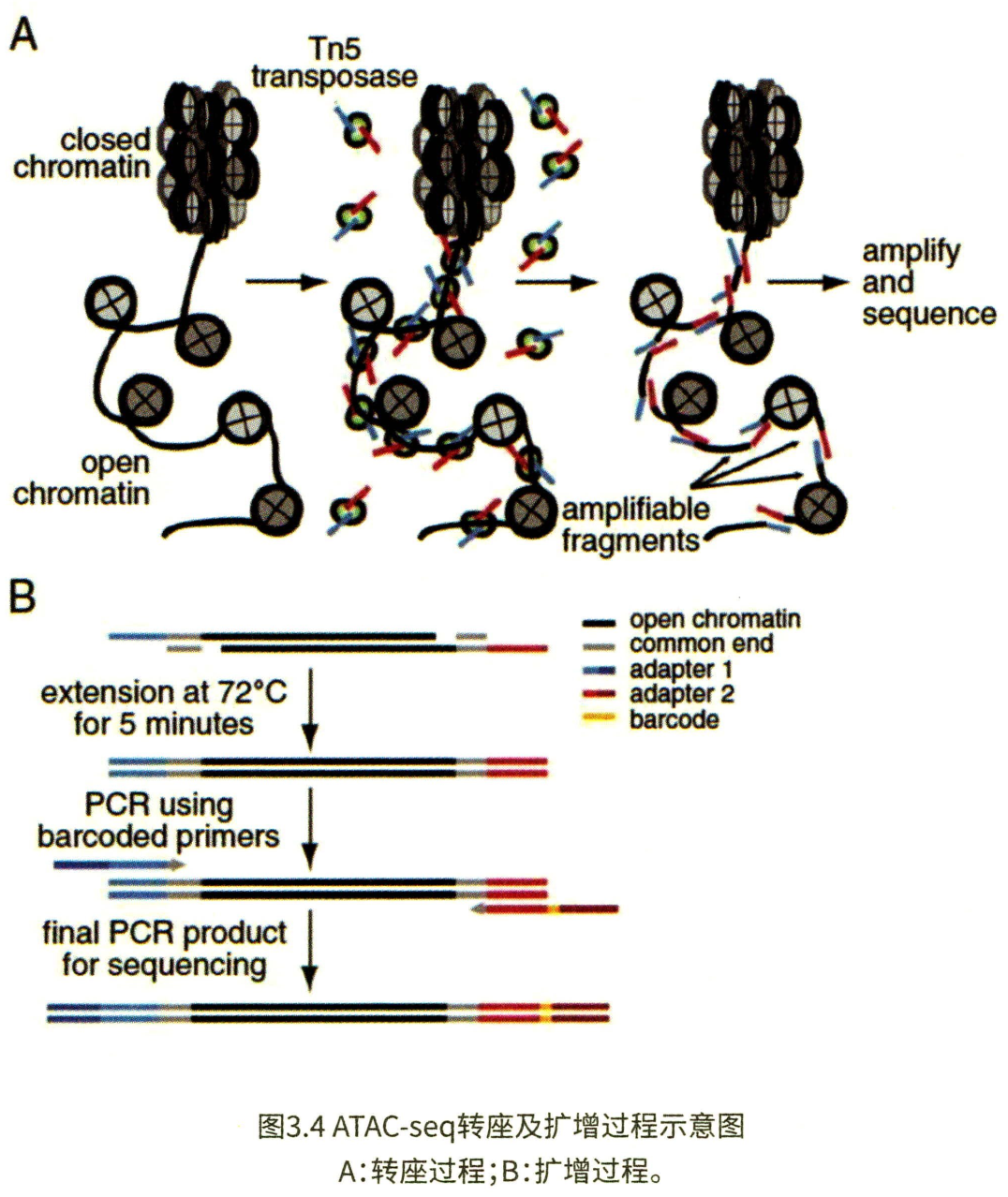
ATAC-seq 实验流程
ATAC-seq 数据质控
ATAC-seq 数据质控流程主要包括预处理和 call peak 两大部分。
首先对数据进行预处理:去接头、测序质控、比对、比对结果过滤、去重,还可以利用 Tn5 酶的切割特性,通过观察插入片段长度的分布判定文库质量。
预处理完成后,进行 peak calling,绘制 reads 在 TSS 附近和整个 genebody 上的分布,计算 TSS 富集分数,进一步判定实验质量。
原始数据 QC 质控及比对
去除 PCR 重复
插入片段长度分布
Reads 在 TSS 附近的分布
Peak Calling
ATAC-seq 高级分析
差异 peak 分析
差异 peak 注释
Motif 富集分析
足迹(footpring)分析
足迹指转录因子(TF)在DNA上的结合位置,由于TF的动态结合,Tn5转座酶切割到次区域的概率低于两侧的完全开放区,因此可通过测序深度的差异识别出此位置,足迹分析的目的在于从数据中鉴别出这些位置。
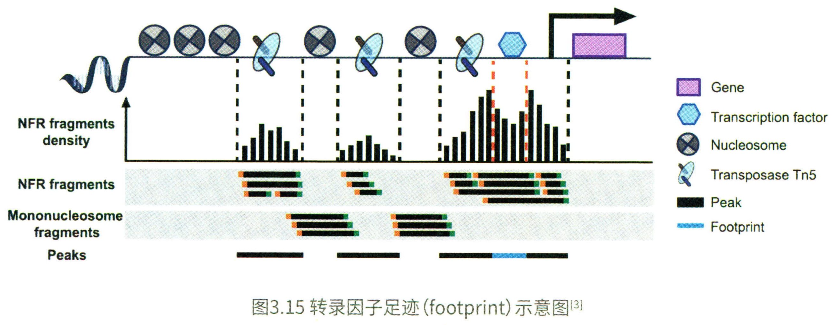
在实践中,我们使用HINT-ATAC进行足迹分析。HINT-ATAC是一种de novo方法,基于HMM模型,并且根据Tn5转座酶的切割特点,对足迹分析的结果进行了校正。Footprint的结果还可以进一步做motif分析。
核小体占位(nucleosome positioning)分析
ATAC 高级分析软件介绍
参考
公众号 | 伯远生物 | 解析表观遗传学的工具——ATAC-seq(一)
公众号 | 伯远生物 | 解析表观遗传学的工具——ATAC-seq(二)

若有收获,就点个赞吧
0 人点赞
