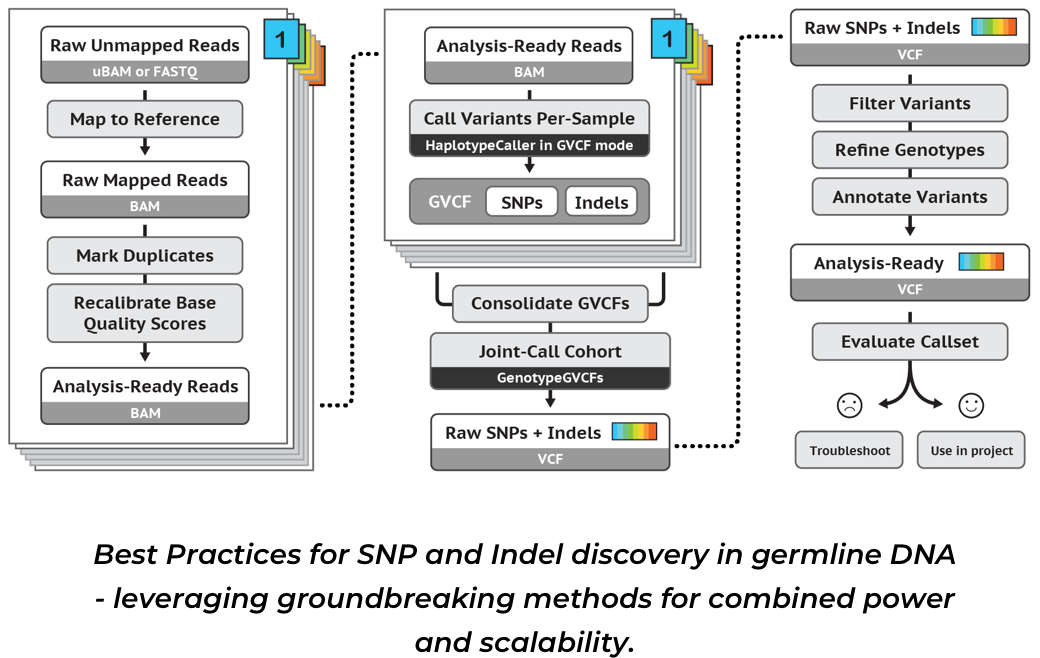
GATK软件介绍
GATK是千人基因组计划中开发出来的进行基因组变异分析的软件。它包含很多命令,其中进行SNP/InDel分析的工具主要有两个:
- UnifiedGenotyper: 基于位点的比对结果分析SNP/InDel。
- HaplotypeCaller: 较新版本才有的命令,取代了UnifiedGenotyper。可以在表现出变异的位点区域使用比对上的reads进行局部的de novo组装,从而能得到更好的SNP/InDel分析结果。还能够很好地处理RNA-Seq的剪接性比对结果,从而可以利用转录组测序数据进行SNP/InDel分析。
HaplotypeCaller
进行SNP/InDel分析的输入文件有两个:
- 参考基因组fasta文件
- 比对到参考基因组上重测序样品的BAM文件(需要@RG信息,可以使用Picard的AddOrReplaceReadGroups添加)
分染色体进行/多染色体进行
# 为什么分染色体进行?gatk --java-options "-Xmx10g -Djava.io.tmpdir=./tmp" HaplotypeCaller -R ../01.ref/genome.fasta -I ../02.mapping/S1.sort.markdup.bam -L chr1 -ERC GVCF -O S1/S1.chr1.g.vcf.gz 1>S1/S1.chr1.HC.log 2>&1
对多个分别使用HaplotypeCaller命令进行分析后,再通过GATK的CombineGVCFs命令将多个样品的文件整合到一起。
gatk --java-options "-Xmx10g -Djava.io.tmpdir=./tmp" CombineGVCFs -R ../01.ref/genome.fasta -V gvcf.chr1.list -O chr1.g.vcf.gz 1>chr1.CombineGVCFs.log 2>&1
GenotypeGVCFs
两种方法
CombineGVCFs
分染色体进行,适合样品较少时使用
GenomicsDB
适合样品较多时使用
额外添加或减少样品时,很方便
gatk --java-options "-Xmx10g -Djava.io.tmpdir=./tmp -DGATK_STACKTRACE_ON_USER_EXCEPTION=true" GenomicsDBImport --sample-name-map gvcf.chr1.map --genomicsdb-workspace-path genomeDB.chr1 -L chr1 --reader-threads 2 --batch-size 50 --tmp-dir ./tmp 1>chr1.GenomicsDBImport.log 2>&1
MergeVcfs
分染色体vcf文件合并
SNP/InDel提取和过滤
使用GATK的VariantFiltration命令对变异位点结果进行过滤。理论上,更好的过滤方法是根据已有的准确的变异位点,通过机器学习的方法对变异位点的质量得分进行矫正。
SNP
InDel
参考
github 地址:https://github.com/broadinstitute/gatk

若有收获,就点个赞吧
0 人点赞
