介绍
ALLHiC 基于 HiC 数据对多倍体基因组进行分型(phasing)和挂载(scaffolding)的软件。
对多倍体基因组进行挂载的主要问题在于,Hi-C 信号经常在等位单倍型之间检测到,现有的任何先进的 Hi-C 挂载程序都会将这些等位单倍型链接在一起。为了解决这个问题,我们开发了一种新的 Hi-C 挂载流程,称为 ALLHIC,专门针对多倍体基因组设计。
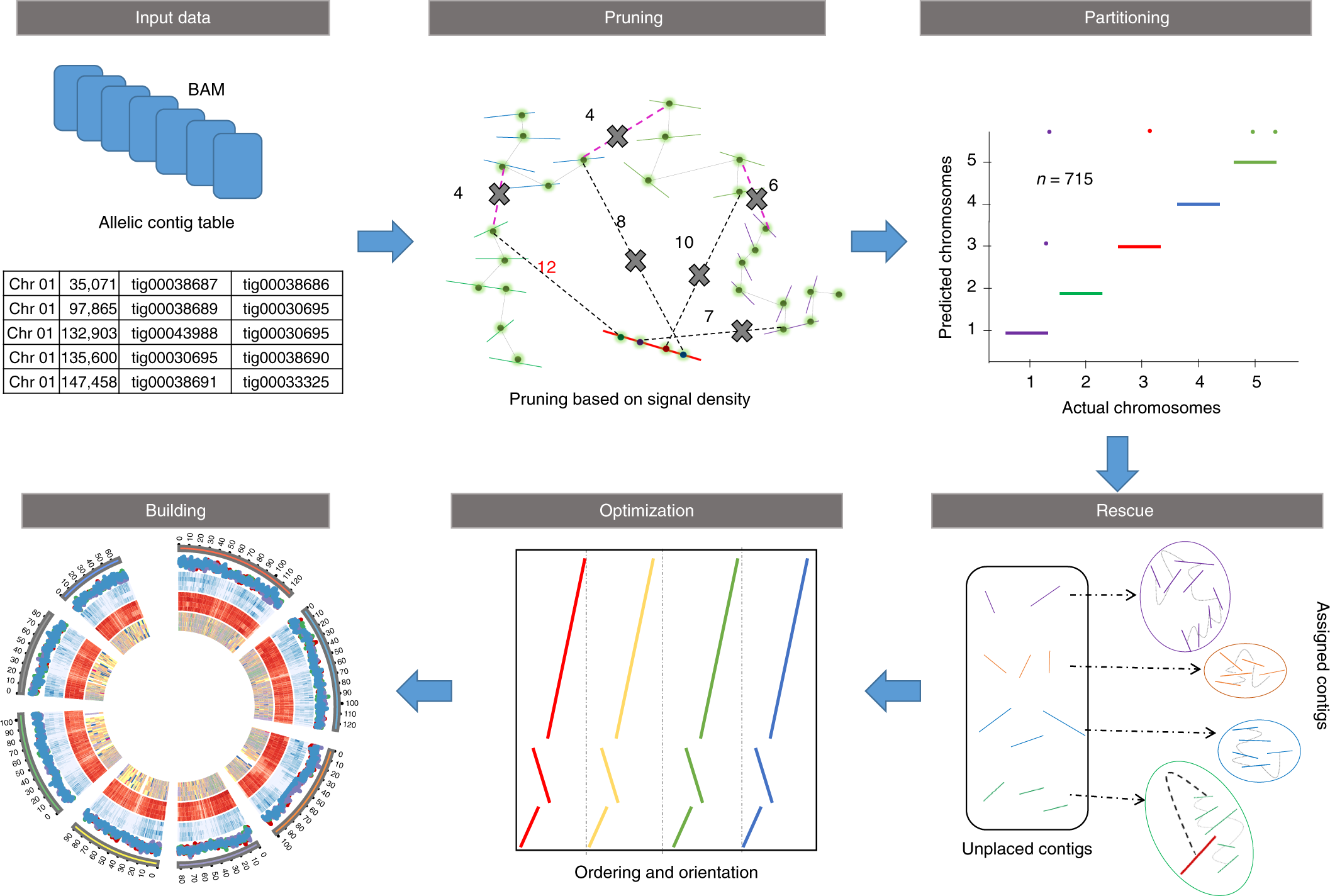
ALLHIC 流程包含五个步骤:
prune: 移除等位基因间的链接,以便更容易单独分离同源染色体;partition: 以剪枝后的bam文件为输入,根据Hi-C建议的链接将链接的contig聚类到预设数量的分区中,假定沿着同一条同源染色体。rescue: 从原始未剪枝的bam文件中搜索未参与分区步骤的contig,并根据Hi-C信号密度将它们分配到特定的集群中。optimize: 对每个分区进行优化,排列所有contig的顺序和方向。build: 通过连接contig、在contig之间添加间隙并生成FASTA格式的最终基因组版本来重建每条染色体。
图1. ALLHiC算法主要步骤概述。新发布的ALLHiC流程包含五个功能:剪枝、分区、救援、优化和构建。简而言之,
ALLHiC2ALLMAPS.pl
ALLHiC_pip.sh
ALLHiC_build
ALLHiC_corrector
ALLHiC_partition
ALLHiC_prune
ALLHiC_rescue
ALLHiC_plot
运行
输入文件准备
bwa index -a bwtsw draft.asm.fastasamtools faidx draft.asm.fasta
bwa aln -t 24 draft.asm.fasta reads_R1.fastq.gz > sample_R1.saibwa aln -t 24 draft.asm.fasta reads_R2.fastq.gz > sample_R2.saibwa sampe draft.asm.fasta sample_R1.sai sample_R2.sai reads_R1.fastq.gz reads_R2.fastq.gz > sample.bwa_aln.sam
PreprocessSAMs.pl sample.bwa_aln.sam draft.asm.fasta MBOI(*)filterBAM_forHiC.pl sample.bwa_aln.REduced.paired_only.bam sample.clean.samsamtools view -bt draft.asm.fasta.fai sample.clean.sam > sample.clean.bam*Tip: skip this step if you are using bwa mem for alignment
prune
剪枝功能首先允许我们检测等位contig,这可以通过基于良好组装的近缘物种或组装好的单倍体基因组来识别共线基因来实现。从输入的BAM文件中移除等位contig之间的信号(归一化的Hi-C读数)。
Usage:ALLHiC_prune -i Allele.ctg.table -b sample.clean.bam -r draft.asm.fastaParameters:-h : help and usage-i : Allele.ctg.table-b : input bam file-r : draft.asm.fastaTips: Details on how to identify allelic contigs can be found in the following link:https://github.com/tangerzhang/ALLHiC/wiki/ALLHiC:-identify-allelic-contigs
https://github.com/tangerzhang/ALLHiC/wiki/ALLHiC:-identify-allelic-contigs
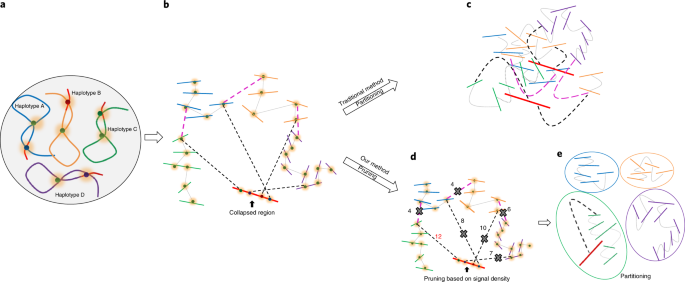
在多倍体基因组组装中,共享高度相似性的单倍型可能会发生折叠。折叠区域与附近分相的单倍型之间的信号会导致嵌合的脚手架。在剪枝步骤中,只保留折叠的contig与分相的contig之间的最佳链接。
图2. 多倍体基因组中Hi-C脚手架问题的描述及剪枝方法在单倍型分相中的应用。
(a) 一个自四倍体基因组的示意图。四条同源染色体分别用不同颜色表示(蓝色、橙色、绿色和紫色)。染色体中的红色区域表示高相似性序列。
(b) 在自四倍体基因组中检测到的Hi-C信号。黑色虚线表示折叠区域与未折叠 contig 之间的 Hi-C 信号。粉红色虚线表示单倍型间的 Hi-C 链接,灰色虚线表示单倍型内的 Hi-C 链接。在组装过程中,由于序列高度相似,红色区域会折叠;而其他区域如果有丰富的变异,则会分离成不同的contig。由于折叠区域在物理上与来自不同单倍型的contig相关联,因此会在折叠区域与所有其他未折叠contig之间检测到Hi-C信号。
(c) 传统的Hi-C脚手架方法会检测来自不同单倍型的contig以及折叠区域之间的信号,并将所有序列聚集在一起。
(d) 剪枝Hi-C信号:1- 移除等位区域之间的信号;2- 只保留折叠区域与未折叠contig之间的最强信号。
(e) 基于剪枝后的Hi-C信息进行分区。理想情况下,contig会根据剪枝结果分相到不同的组。
prune 步骤去除了等位基因之间的联系,因此同源染色体更易于单独分离。
partition
partition 功能将修剪的bam文件作为输入,并根据Hi-C建议的链接对链接的contigs进行聚类,大概是沿着相同同源染色体在预设数量的分区中进行。
rescue
rescue 功能从原始未修剪的bam文件中搜索分区步骤中不涉及的contigs,并根据Hi-C信号密度将它们分配给特定的群集。
optimize
optimize 步骤采用每个分区,并优化所有contigs的顺序和方向。
build
build 步骤通过连接contigs来重建每个染色体
参考
github 地址:https://github.com/tangerzhang/ALLHiC
Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data

若有收获,就点个赞吧
0 人点赞
