基本分析
在本节中,我们将使用immunarch包中的基本函数对免疫组库数据进行常规分析。
常用函数如下:
repExplore- 计算基本统计数据,例如克隆数或长度和计数的分布。要探索它们,您需要将统计信息如“count”传递给.method参数.repClonality- 计算TCR/BCR库的克隆性。repOverlap- 计算不同TCR/BCR库的重叠性。repOverlapAnalysis- 分析TCR/BCR库的重叠,包括不同的聚类程序和 PCA。geneUsage- 计算V或J基因的分布。geneUsageAnalysis- 分析V或J基因的分布,包括聚类和PCA。repDiversity- 估计TCR/BCR库的多样性。trackClonotypes- 分析跨时间点的TCR/BCR库动态。spectratype- 计算克隆型的谱型。getKmers和kmer_profile- 计算 kmers 和序列配置文件的分布。
可视化分析结果
在immunarch中,每个函数的分析结果都可以直接传递给vis函数进行可视化,它是一个通用的可视化函数。同时,我们还可以通过.by参数传递任一分组变量,或同时将.by和.meta参数传递给 vis函数来进行分组展示。
- 我们可以通过
.by参数将元数据表中的一个或多个列名称作为字符向量传递,以便在绘图之前对数据进行分组。在这种情况下,您还应该提供.meta元数据表的参数。
- 我们可以通过
exp_vol <- repExplore(immdata$data, .method = "volume")# Sample Volume# A2-i129 A2-i129 6443# A2-i131 A2-i131 6375# A2-i133 A2-i133 6300# A2-i132 A2-i132 6721# A4-i191 A4-i191 5058# A4-i192 A4-i192 5763p1 <- vis(exp_vol, .by = c("Status"), .meta = immdata$meta)p2 <- vis(exp_vol, .by = c("Status", "Sex"), .meta = immdata$meta)# p1 + p2
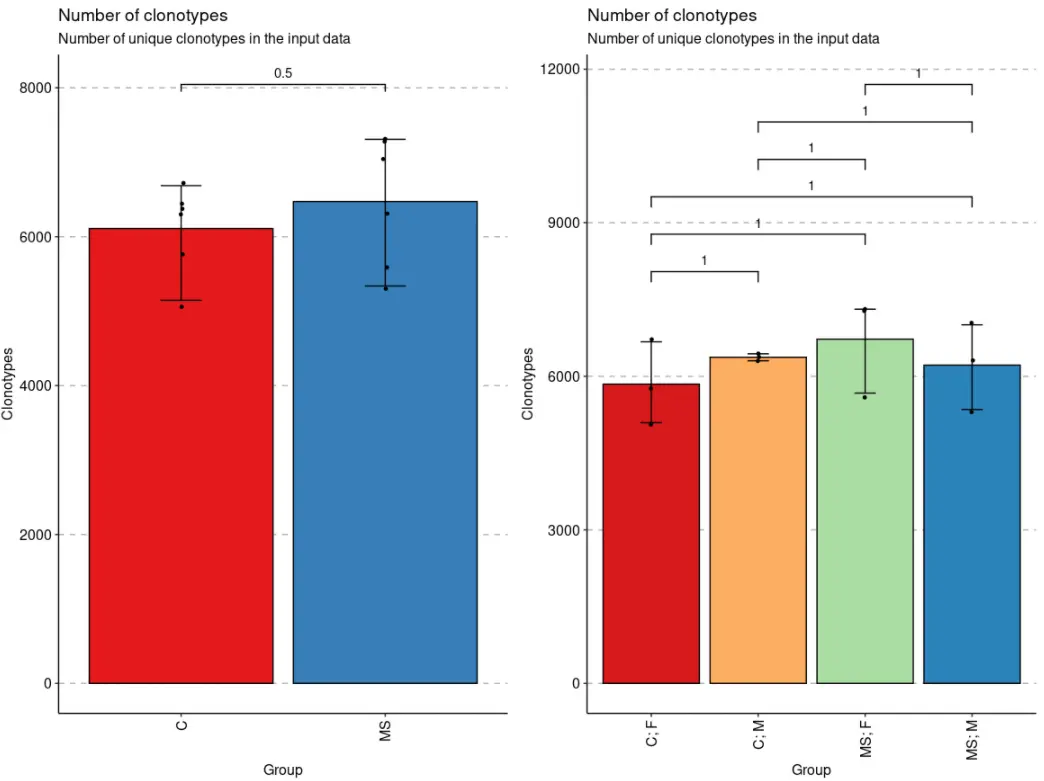
- 我们还可以将
.by字符向量作为与数据中样本数量完全匹配的字符向量传递,每个值应对应于一个样本的属性。它将用于根据提供的值对数据进行分组。请注意,在这种情况下,您应该将 NA 传递给.meta.
- 我们还可以将
exp_vol <- repExplore(immdata$data, .method = "volume")by_vec <- c("C", "C", "C", "C", "C", "C", "MS", "MS", "MS", "MS", "MS", "MS")p <- vis(exp_vol, .by = by_vec)p
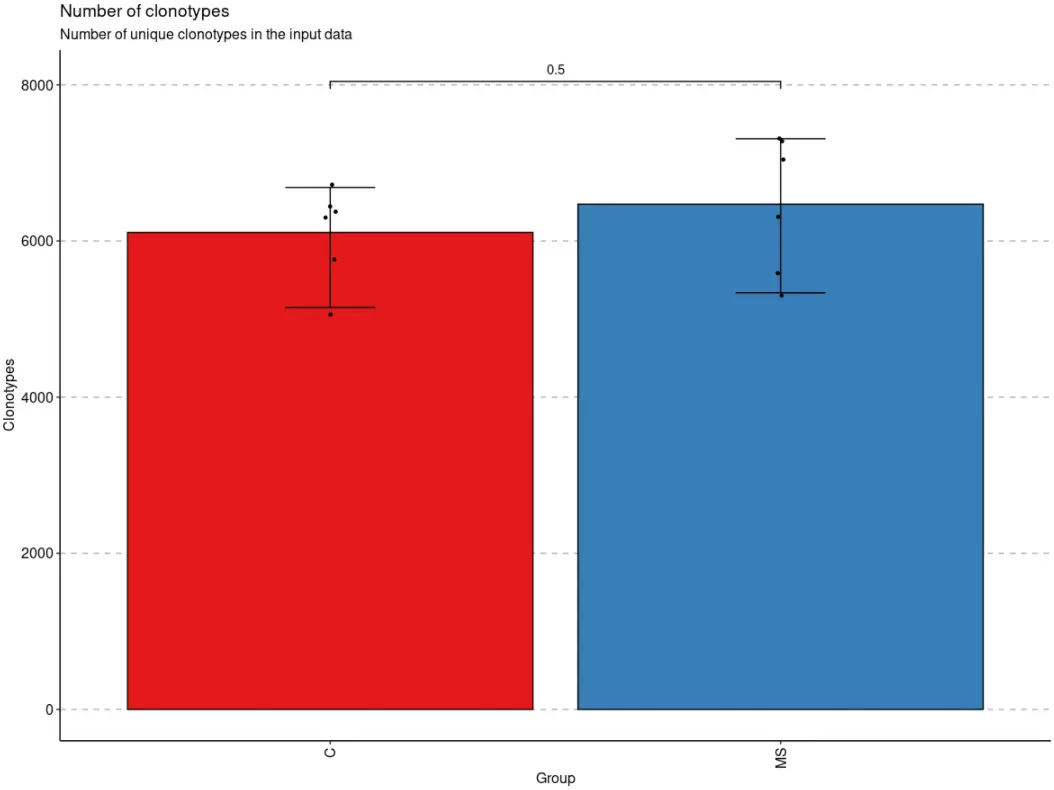
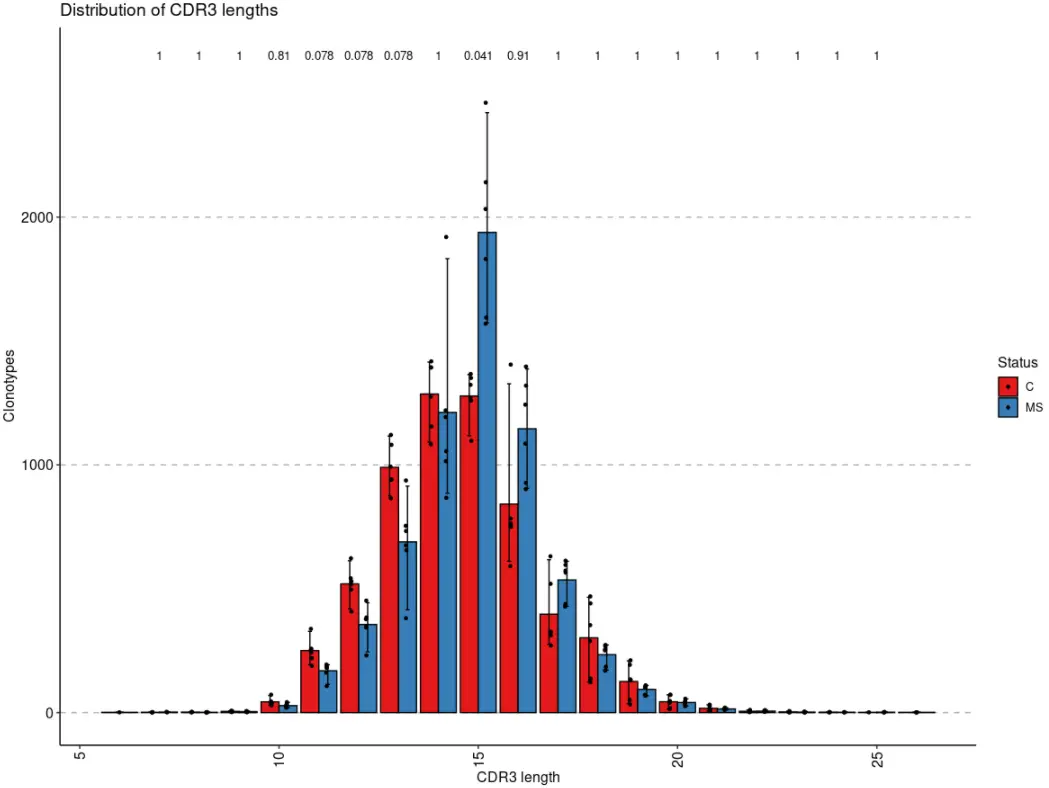
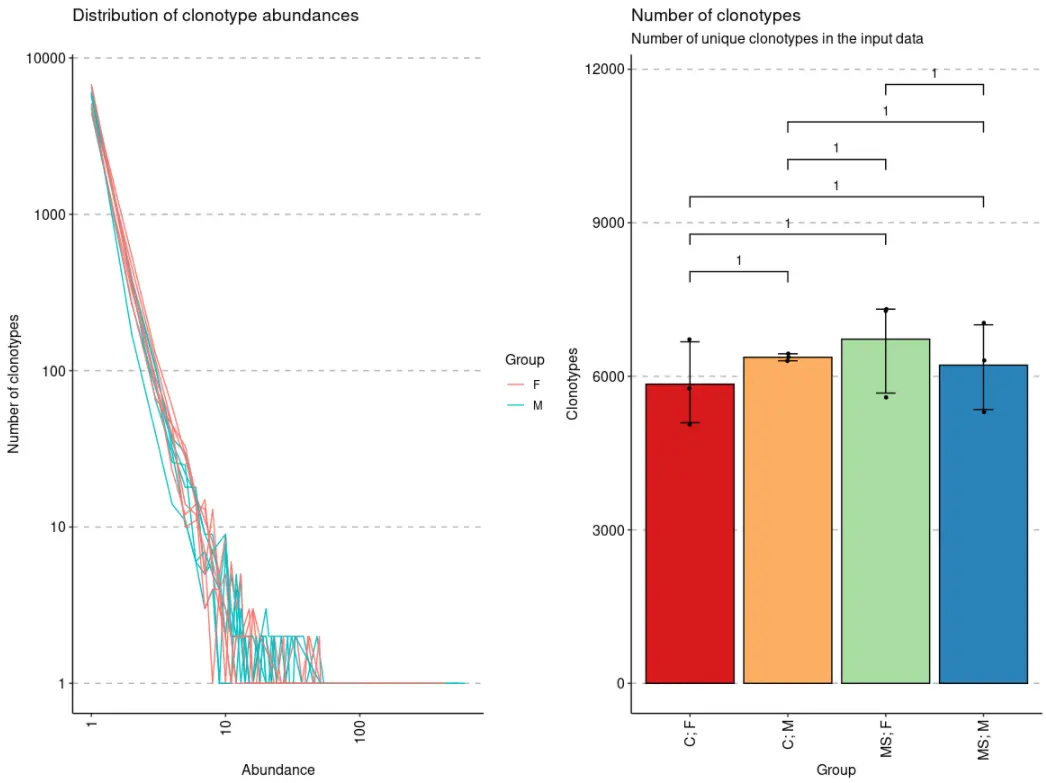
当数据被分组后,immunarch包会默认执行组间均值比较的统计检验,除非设置参数.test = F。如果只有两个组,则执行Wilcoxon 秩和检验(wilcox.test带有参数的R 函数exact = F)以测试两组之间的平均秩值是否存在差异。如果有两个以上的组,则执行Kruskal-Wallis 检验(R 函数kruskal.test),这相当于秩合方差分析,它测试来自不同组的样本是否来自同一分布。一个显著的 Kruskal-Wallis 检验结果表明至少一个样本随机地支配另一个样本。多重比较调整后的 P 值绘制在组的顶部。P 值调整是使用Holm 方法(也称为 Holm-Bonferroni 校正)。您可以[?p.adjust](https://rdrr.io/r/stats/p.adjust.html)在 R 控制台中执行该命令以查看更多信息。
该vis函数生成的图以及任何基于 ggplot2 的图都可以传递给 —fixVis内置交互式可视化工具,用于制作可发表的图:
# 1. Analyseexp_len <- repExplore(immdata$data, .method = "len", .col = "aa")# Sample Length Count# 1 A2-i129 6 1# 2 A2-i129 9 6# 3 A2-i129 10 36# 4 A2-i129 11 243# 5 A2-i129 12 517# 6 A2-i129 13 1081# 2. Visualisep1 <- vis(exp_len)# 3. Fix and make publication-ready resultsfixVis(p1)
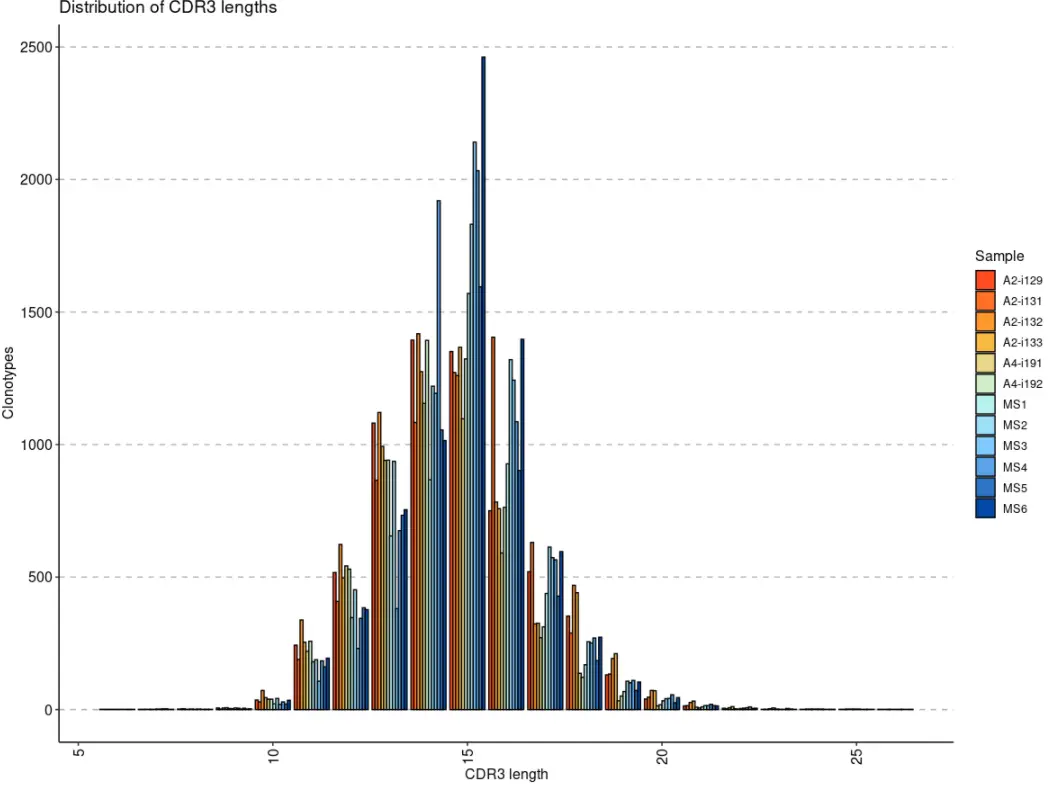
探索性可视化分析
对于基本的探索性分析,例如比较每个曲目的读取数/UMI 或分布,请使用该函数repExplore。
exp_len <- repExplore(immdata$data, .method = "len", .col = "aa")head(exp_len)# Sample Length Count# 1 A2-i129 6 1# 2 A2-i129 9 6# 3 A2-i129 10 36# 4 A2-i129 11 243# 5 A2-i129 12 517# 6 A2-i129 13 1081exp_cnt <- repExplore(immdata$data, .method = "count")head(exp_cnt)# Sample Clone.num Clonotypes# 1 A2-i129 1 5858# 2 A2-i129 2 388# 3 A2-i129 3 86# 4 A2-i129 4 26# 5 A2-i129 5 25# 6 A2-i129 6 6exp_vol <- repExplore(immdata$data, .method = "volume")head(exp_vol)# Sample Volume# A2-i129 A2-i129 6443# A2-i131 A2-i131 6375# A2-i133 A2-i133 6300# A2-i132 A2-i132 6721# A4-i191 A4-i191 5058# A4-i192 A4-i192 5763p1 <- vis(exp_len)p2 <- vis(exp_cnt)p3 <- vis(exp_vol)p1
p2 + p3
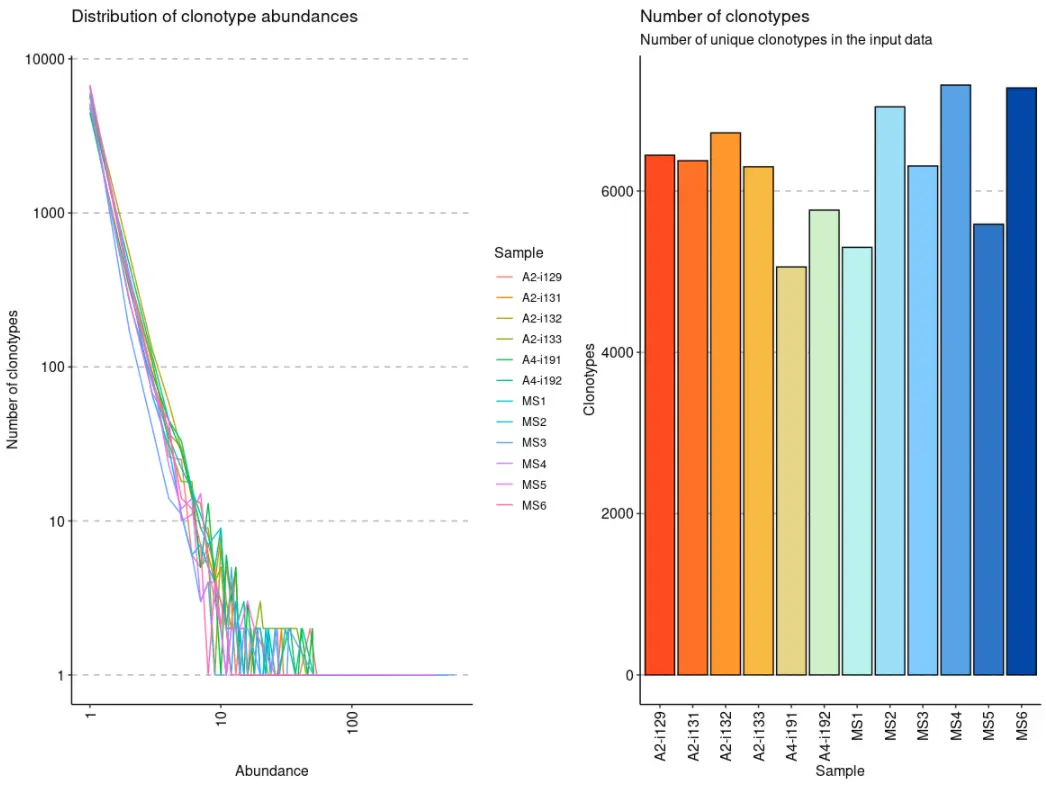
# You can group samples by their metainformationp4 <- vis(exp_len, .by = "Status", .meta = immdata$meta)p5 <- vis(exp_cnt, .by = "Sex", .meta = immdata$meta)p6 <- vis(exp_vol, .by = c("Status", "Sex"), .meta = immdata$meta)p4
p5 + p6
克隆性分析
估计样本多样性的方法之一是评估克隆性。repClonality函数可以计算最频繁或最不常见的克隆型的数量。有几种方法可以评估克隆性,让我们来看看它们。该clonal.prop方法计算细胞克隆池占据的所有库的比例:
imm_pr <- repClonality ( immdata$data , .method = "clonal.prop" )imm_pr## Clones Percentage Clonal.count.prop## A2-i129 18 10.0 0.0027556644## A2-i131 28 10.0 0.0042728521## A2-i133 9 10.3 0.0014077898## A2-i132 113 10.0 0.0164987589## A4-i191 4 11.5 0.0007773028## A4-i192 8 10.4 0.0013738623## MS1 2 10.1 0.0003700278## MS2 66 10.0 0.0092372288## MS3 2 10.6 0.0003095496## MS4 176 10.0 0.0236336780## MS5 3 13.2 0.0005303164## MS6 150 10.0 0.0202456472## attr(,"class")## [1] "immunr_clonal_prop" "matrix"
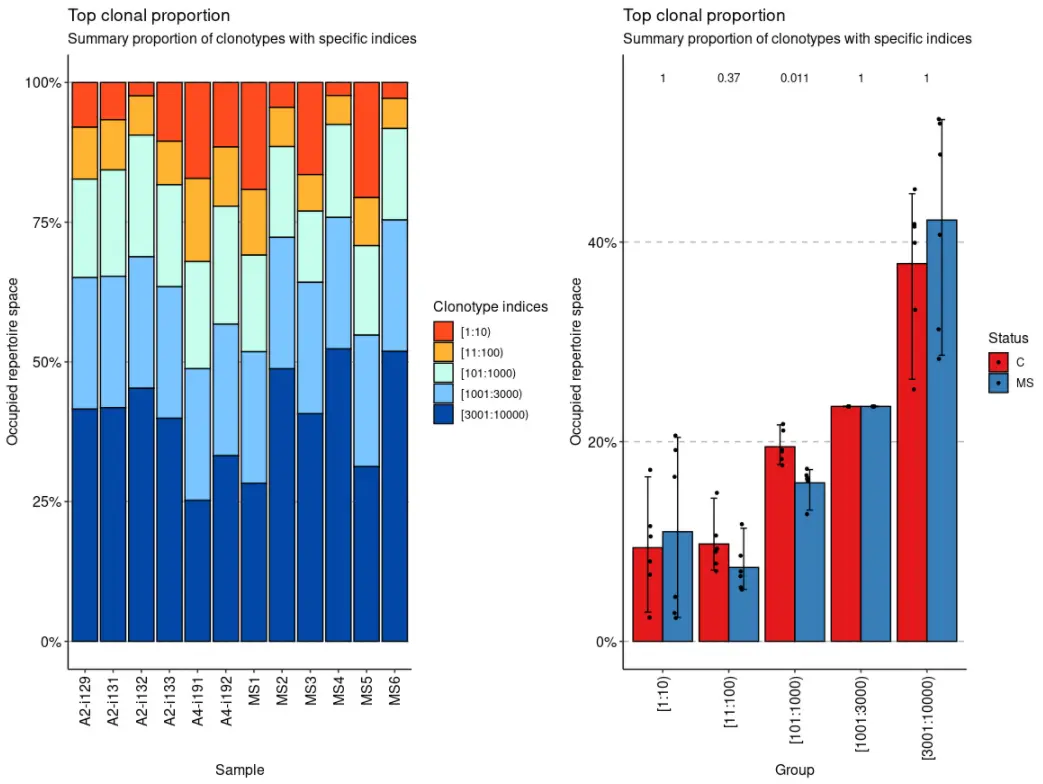
该top方法考虑了最丰富的细胞克隆型:
imm_top <- repClonality(immdata$data, .method = "top", .head = c(10, 100, 1000, 3000, 10000))imm_top## 10 100 1000 3000 10000## A2-i129 0.08011765 0.17282353 0.3491765 0.5844706 1## A2-i131 0.06670588 0.15647059 0.3467059 0.5820000 1## A2-i133 0.10505882 0.18294118 0.3655294 0.6008235 1## A2-i132 0.02388235 0.09423529 0.3118824 0.5471765 1## A4-i191 0.17176471 0.32047059 0.5122353 0.7475294 1## A4-i192 0.11541176 0.22141176 0.4325882 0.6678824 1## MS1 0.19164706 0.30894118 0.4817647 0.7170588 1## MS2 0.04458824 0.11470588 0.2770588 0.5123529 1## MS3 0.16482353 0.23011765 0.3575294 0.5928235 1## MS4 0.02329412 0.07517647 0.2415294 0.4768235 1## MS5 0.20611765 0.29188235 0.4521176 0.6874118 1## MS6 0.02835294 0.08235294 0.2460000 0.4812941 1## attr(,"class")## [1] "immunr_top_prop" "matrix"
该rare方法处理丰度最低的克隆型:
imm_rare <- repClonality(immdata$data, .method = "rare")imm_rare## 1 3 10 30 100 MAX## A2-i129 0.6991765 0.8215294 0.8710588 0.9267059 0.9604706 1## A2-i131 0.6969412 0.8243529 0.8995294 0.9436471 0.9869412 1## A2-i133 0.6800000 0.8100000 0.8690588 0.8974118 0.9357647 1## A2-i132 0.7088235 0.8831765 0.9643529 0.9951765 1.0000000 1## A4-i191 0.5355294 0.6484706 0.7189412 0.7707059 0.8697647 1## A4-i192 0.5976471 0.7487059 0.8342353 0.8675294 0.9156471 1## MS1 0.5780000 0.6692941 0.7438824 0.7929412 0.8571765 1## MS2 0.7788235 0.8891765 0.9322353 0.9718824 1.0000000 1## MS3 0.7272941 0.7825882 0.8071765 0.8385882 0.8652941 1## MS4 0.8109412 0.9343529 0.9725882 1.0000000 1.0000000 1## MS5 0.6112941 0.7001176 0.7575294 0.7809412 0.8284706 1## MS6 0.8107059 0.9208235 0.9703529 0.9897647 1.0000000 1## attr(,"class")## [1] "immunr_rare_prop" "matrix"
最后,该homeo方法评估克隆空间稳态,即给定大小的克隆占据的TCR/BCR库的比例:
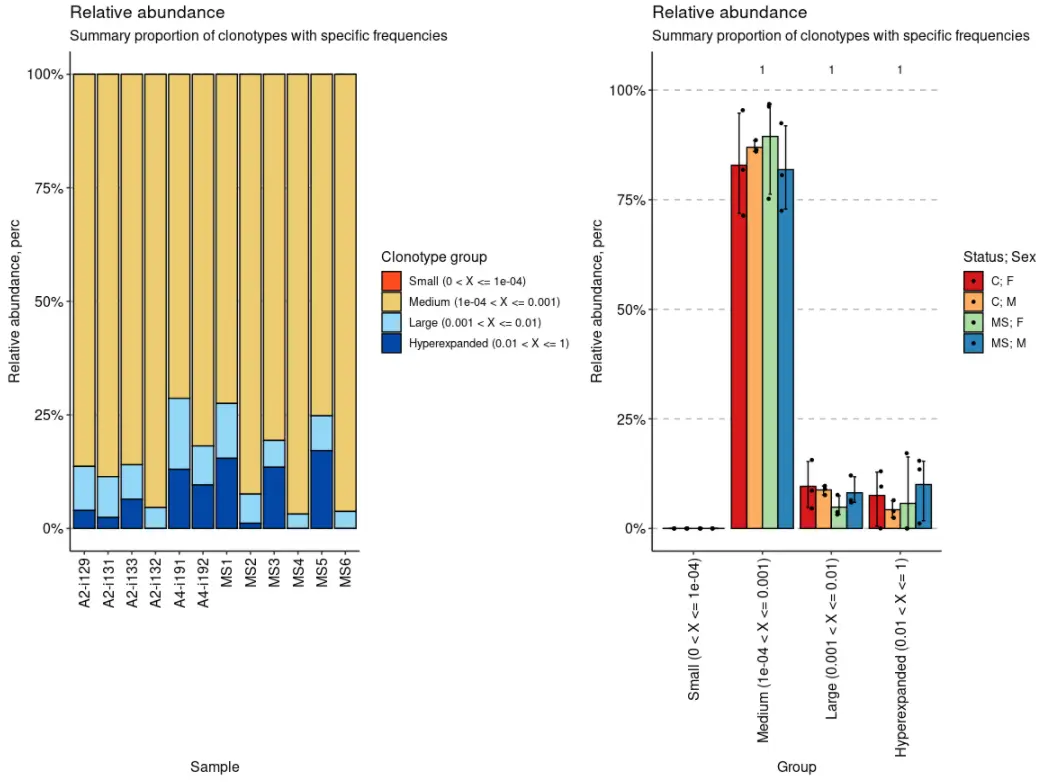
imm_hom <- repClonality(immdata$data,.method = "homeo",.clone.types = c(Small = .0001, Medium = .001, Large = .01, Hyperexpanded = 1))imm_hom## Small (0 < X <= 1e-04) Medium (1e-04 < X <= 0.001)## A2-i129 0 0.8634118## A2-i131 0 0.8858824## A2-i133 0 0.8597647## A2-i132 0 0.9542353## A4-i191 0 0.7135294## A4-i192 0 0.8183529## MS1 0 0.7248235## MS2 0 0.9244706## MS3 0 0.8061176## MS4 0 0.9683529## MS5 0 0.7520000## MS6 0 0.9625882## Large (0.001 < X <= 0.01) Hyperexpanded (0.01 < X <= 1)## A2-i129 0.09705882 0.03952941## A2-i131 0.09011765 0.02400000## A2-i133 0.07600000 0.06423529## A2-i132 0.04576471 0.00000000## A4-i191 0.15623529 0.13023529## A4-i192 0.08611765 0.09552941## MS1 0.12082353 0.15435294## MS2 0.06411765 0.01141176## MS3 0.05917647 0.13470588## MS4 0.03164706 0.00000000## MS5 0.07647059 0.17152941## MS6 0.03741176 0.00000000## attr(,"class")## [1] "immunr_homeo" "matrix"
vis(imm_top) + vis(imm_top, .by = "Status", .meta = immdata$meta)
vis(imm_rare) + vis(imm_rare, .by = "Status", .meta = immdata$meta)
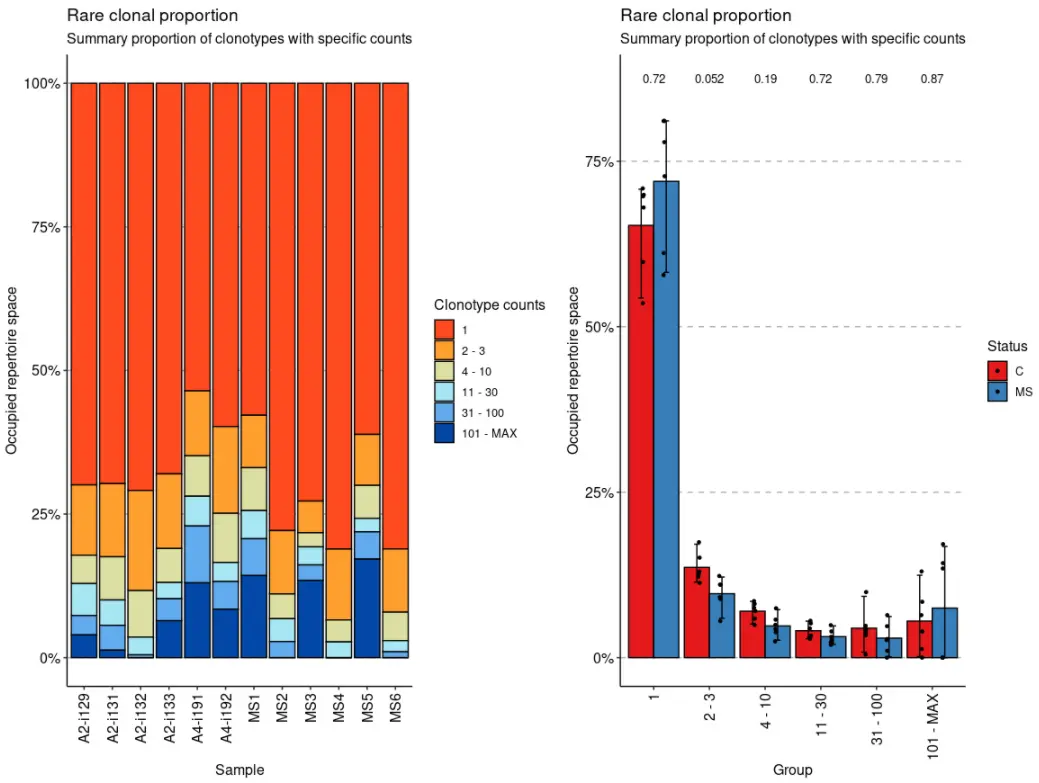
vis(imm_hom) + vis(imm_hom, .by = c("Status", "Sex"), .meta = immdata$meta)
参考来源:https://immunarch.com/articles/web_only/v3_basic_analysis.html

若有收获,就点个赞吧
0 人点赞
