Seurat3引入了用于多个单细胞测序数据集进行整合分析的新方法。这些方法可以对来自不同的个体、实验条件、测序技术甚至物种中收集来的数据进行整合,旨在识别出不同数据集之间的共享细胞状态(shared cell states)。
这些方法首先识别出不同数据集对之间的“锚(anchors)”,这些anchors代表了个体细胞之间成对的对应关系(每个数据集中有一个),并假设它们源自相同的生物状态。然后,再利用这些识别出的anchors用于协调不同的数据集,或者将信息从一个数据集传输到另一个数据集。
一、标准工作流程进行整合分析
在本例教程中,我们选择了通过四种不同测序技术(CelSeq (GSE81076)、 CelSeq2 (GSE85241)、Fluidigm C1 (GSE86469)和SMART-Seq2 (E-MTAB-5061)生成的人类胰岛细胞数据集,我们通过SeuratData包来加载这个数据集。
安装并加载所需的R包
# 安装并加载SeuratData包devtools::install_github('satijalab/seurat-data')library(SeuratData)library(Seurat)# 查看SeuratData包搜集的数据集AvailableData()Dataset Version Summary species system ncells tech notes Installed InstalledVersioncbmc.SeuratData cbmc 3.0.0 scRNAseq and 13-antibody sequencing of CBMCs human CBMC (cord blood) 8617 CITE-seq <NA> TRUE 3.0.0hcabm40k.SeuratData hcabm40k 3.0.0 40,000 Cells From the Human Cell Atlas ICA Bone Marrow Dataset human bone marrow 40000 10x v2 <NA> FALSE 3.0.0ifnb.SeuratData ifnb 3.0.0 IFNB-Stimulated and Control PBMCs human PBMC 13999 10x v1 <NA> TRUE 3.0.0panc8.SeuratData panc8 3.0.0 Eight Pancreas Datasets Across Five Technologies human Pancreatic Islets 14892 SMARTSeq2, Fluidigm C1, CelSeq, CelSeq2, inDrops <NA> TRUE 3.0.0pbmc3k.SeuratData pbmc3k 3.0.0 3k PBMCs from 10X Genomics human PBMC 2700 10x v1 <NA> TRUE 3.0.0pbmcsca.SeuratData pbmcsca 3.0.0 Broad Institute PBMC Systematic Comparative Analysis human PBMC 31021 10x v2, 10x v3, SMARTSeq2, Seq-Well, inDrops, Drop-seq, CelSeq2 HCA benchmark FALSE 3.0.0# 下载安装SeuratData包收集的特定数据集InstallData("panc8")# 加载数据集library(panc8.SeuratData)data("panc8")panc8An object of class Seurat34363 features across 14890 samples within 1 assayActive assay: RNA (34363 features)
分割对象,构建不同的数据集
head(panc8@meta.data)orig.ident nCount_RNA nFeature_RNA tech replicate assigned_clusterD101_5 D101 4615.810 1986 celseq celseq <NA>D101_7 D101 29001.563 4209 celseq celseq <NA>D101_10 D101 6707.857 2408 celseq celseq <NA>D101_13 D101 8797.224 2964 celseq celseq <NA>D101_14 D101 5032.558 2264 celseq celseq <NA>D101_17 D101 13474.866 3982 celseq celseq <NA>celltype datasetD101_5 gamma celseqD101_7 acinar celseqD101_10 alpha celseqD101_13 delta celseqD101_14 beta celseqD101_17 ductal celseq# 根据meta信息中不同的测序技术(tech)对Seurat对象进行分割,构建不同的数据集pancreas.list <- SplitObject(panc8, split.by = "tech")# 选择出四种不同测序技术产生的数据pancreas.list <- pancreas.list[c("celseq", "celseq2", "fluidigmc1", "smartseq2")]pancreas.list$celseqAn object of class Seurat34363 features across 1004 samples within 1 assayActive assay: RNA (34363 features, 0 variable features)$celseq2An object of class Seurat34363 features across 2285 samples within 1 assayActive assay: RNA (34363 features, 0 variable features)$fluidigmc1An object of class Seurat34363 features across 638 samples within 1 assayActive assay: RNA (34363 features, 0 variable features)$smartseq2An object of class Seurat34363 features across 2394 samples within 1 assayActive assay: RNA (34363 features, 0 variable features)
分别对每个数据集进行标准的预处理
for (i in 1:length(pancreas.list)) {pancreas.list[[i]] <- NormalizeData(pancreas.list[[i]], verbose = FALSE)pancreas.list[[i]] <- FindVariableFeatures(pancreas.list[[i]], selection.method = "vst", nfeatures = 2000, verbose = FALSE)}
将不同的数据集进行整合
首先使用FindIntegrationAnchors函数来识别anchors,该函数接受Seurat对象的列表(list)作为输入,在这里我们将三个对象构建成一个参考数据集。使用默认参数来识别锚,如数据集的“维数”(30)
reference.list <- pancreas.list[c("celseq", "celseq2", "smartseq2")]pancreas.anchors <- FindIntegrationAnchors(object.list = reference.list, dims = 1:30)Computing 2000 integration featuresScaling features for provided objects|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed = 02sFinding all pairwise anchors| | 0 % ~calculating Running CCAMerging objectsFinding neighborhoodsFinding anchorsFound 3514 anchorsFiltering anchorsRetained 2761 anchorsExtracting within-dataset neighbors|+++++++++++++++++ | 33% ~25s Running CCAMerging objectsFinding neighborhoodsFinding anchorsFound 3500 anchorsFiltering anchorsRetained 2728 anchorsExtracting within-dataset neighbors|++++++++++++++++++++++++++++++++++ | 67% ~12s Running CCAMerging objectsFinding neighborhoodsFinding anchorsFound 6174 anchorsFiltering anchorsRetained 4561 anchorsExtracting within-dataset neighbors|++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed = 49spancreas.anchorsAn AnchorSet object containing 20100 anchors between 3 Seurat objectsThis can be used as input to IntegrateData or TransferData.
然后将这些识别好的anchors传递给IntegrateData函数,整合后的数据返回一个Seurat对象,该对象中将包含一个新的Assay(integrated),里面存储了整合后表达矩阵,原始的表达矩阵存储在RNA这个Assay中。
pancreas.integrated <- IntegrateData(anchorset = pancreas.anchors, dims = 1:30)Merging dataset 1 into 2Extracting anchors for merged samplesFinding integration vectorsFinding integration vector weights0% 10 20 30 40 50 60 70 80 90 100%[----|----|----|----|----|----|----|----|----|----|**************************************************|Integrating dataMerging dataset 3 into 2 1Extracting anchors for merged samplesFinding integration vectorsFinding integration vector weights0% 10 20 30 40 50 60 70 80 90 100%[----|----|----|----|----|----|----|----|----|----|**************************************************|Integrating datapancreas.integratedAn object of class Seurat36363 features across 5683 samples within 2 assaysActive assay: integrated (2000 features, 2000 variable features)1 other assay present: RNA
对整合后的数据集进行常规的降维聚类可视化
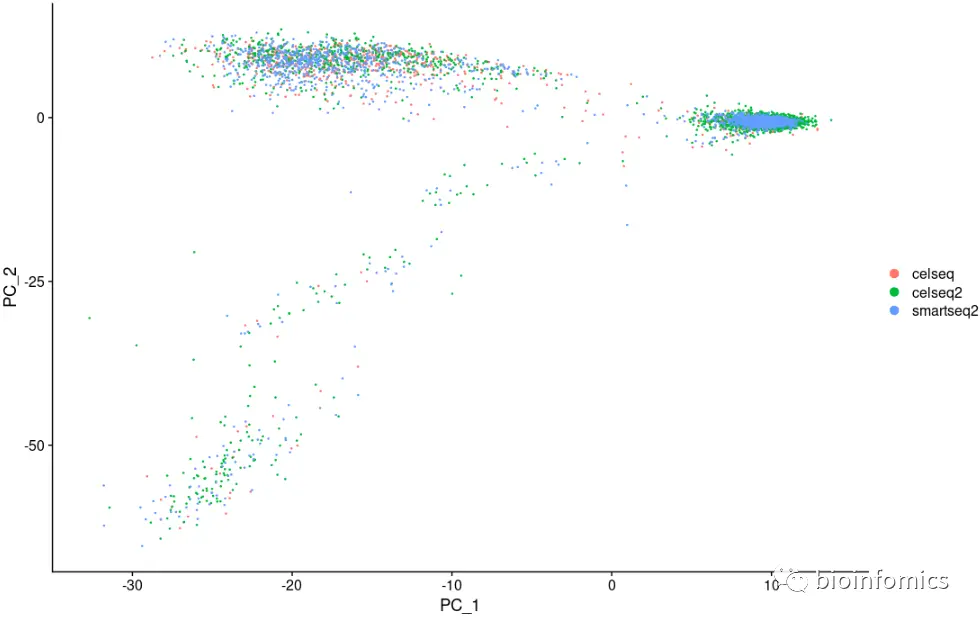
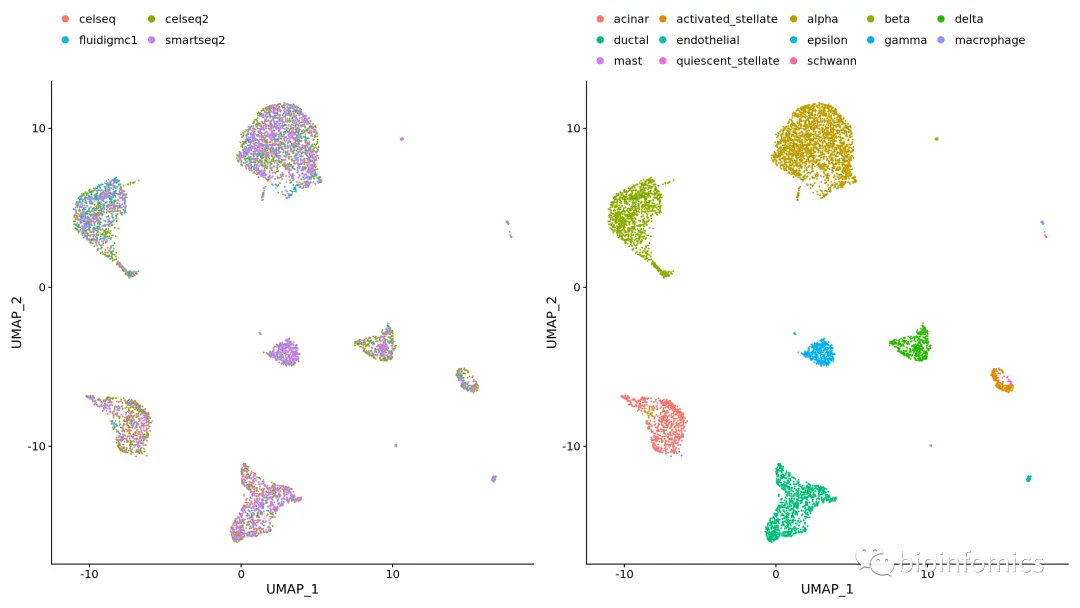
library(ggplot2)library(cowplot)library(patchwork)# switch to integrated assay. The variable features of this assay are automatically# set during IntegrateDataDefaultAssay(pancreas.integrated) <- "integrated"# Run the standard workflow for visualization and clustering# 数据标准化pancreas.integrated <- ScaleData(pancreas.integrated, verbose = FALSE)# PCA降维pancreas.integrated <- RunPCA(pancreas.integrated, npcs = 30, verbose = FALSE)DimPlot(pancreas.integrated, reduction = "pca", group.by = "tech")
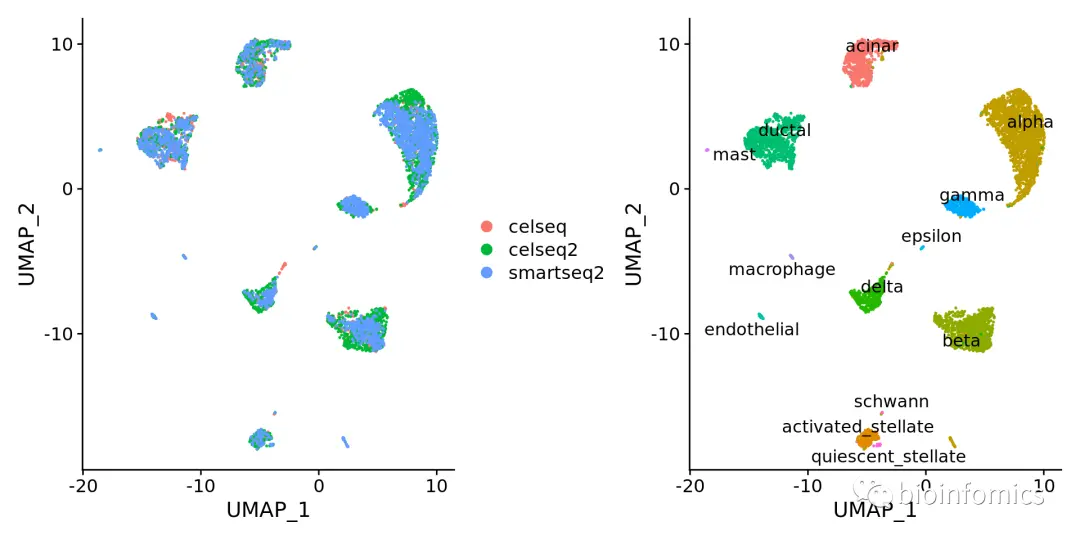
# UMAP降维可视化pancreas.integrated <- RunUMAP(pancreas.integrated, reduction = "pca", dims = 1:30)19:12:41 UMAP embedding parameters a = 0.9922 b = 1.11219:12:41 Read 5683 rows and found 30 numeric columns19:12:41 Using Annoy for neighbor search, n_neighbors = 3019:12:41 Building Annoy index with metric = cosine, n_trees = 500% 10 20 30 40 50 60 70 80 90 100%[----|----|----|----|----|----|----|----|----|----|**************************************************|19:12:43 Writing NN index file to temp file /tmp/RtmpwSZSgF/file2786475d2789719:12:43 Searching Annoy index using 1 thread, search_k = 300019:12:45 Annoy recall = 100%19:12:46 Commencing smooth kNN distance calibration using 1 thread19:12:49 Found 2 connected components, falling back to 'spca' initialization with init_sdev = 119:12:49 Initializing from PCA19:12:49 PCA: 2 components explained 44.22% variance19:12:49 Commencing optimization for 500 epochs, with 252460 positive edges0% 10 20 30 40 50 60 70 80 90 100%[----|----|----|----|----|----|----|----|----|----|**************************************************|19:13:07 Optimization finished# 使用group.by函数根据不同的条件进行分群p1 <- DimPlot(pancreas.integrated, reduction = "umap", group.by = "tech")p2 <- DimPlot(pancreas.integrated, reduction = "umap", group.by = "celltype", label = TRUE, repel = TRUE) + NoLegend()p1 + p2
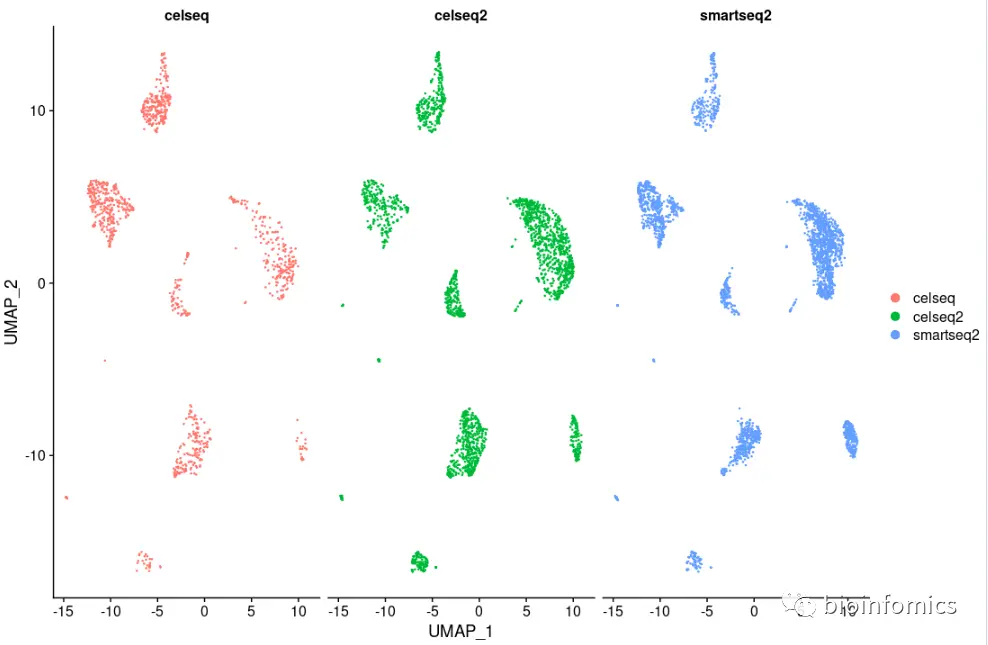
p3 <- DimPlot(pancreas.integrated, reduction = "umap", split.by = "tech")p3
使用整合后的参考数据集对细胞类型进行分类
Seurat3还支持将参考数据集(或元数据)投影到查询对象上。虽然许多方法是一致的(这两个过程都是从识别锚开始的),但数据映射(data transfer)和数据整合(data integration)之间有两个重要的区别:
1)In data transfer, Seurat does not correct or modify the query expression data.
2)In data transfer, Seurat has an option (set by default) to project the PCA structure of a reference onto the query, instead of learning a joint structure with CCA. We generally suggest using this option when projecting data between scRNA-seq datasets.
识别到anchors之后,我们使用TransferData函数根据参考数据集中细胞类型标签向量对查询数据集的细胞进行分类。TransferData函数返回一个带有预测id和预测分数的矩阵,我们可以将其添加到query metadata中。
# 构建query数据集pancreas.query <- pancreas.list[["fluidigmc1"]]pancreas.queryAn object of class Seurat34363 features across 638 samples within 1 assayActive assay: RNA (34363 features, 2000 variable features)# 识别参考数据集的anchorspancreas.anchors <- FindTransferAnchors(reference = pancreas.integrated, query = pancreas.query, dims = 1:30)pancreas.anchorsAn AnchorSet object containing 20100 anchors between 3 Seurat objectsThis can be used as input to IntegrateData or TransferData.# 将查询数据集映射到参考数据集上predictions <- TransferData(anchorset = pancreas.anchors, refdata = pancreas.integrated$celltype, dims = 1:30)# 添加预测出的信息pancreas.query <- AddMetaData(pancreas.query, metadata = predictions)
因为我们具有来自整合后数据集中含有的原始注释标签,所以我们可以评估预测的细胞类型注释与完整参考的匹配程度。在此示例中,我们发现在细胞类型分类中具有很高的一致性,有超过97%的细胞被正确的标记出。
pancreas.query$prediction.match <- pancreas.query$predicted.id == pancreas.query$celltypetable(pancreas.query$prediction.match)## FALSE TRUE## 18 620
为了进一步验证这一点,我们可以查看一些特定胰岛细胞群中的典型细胞类型标记基因(cell type markers)。
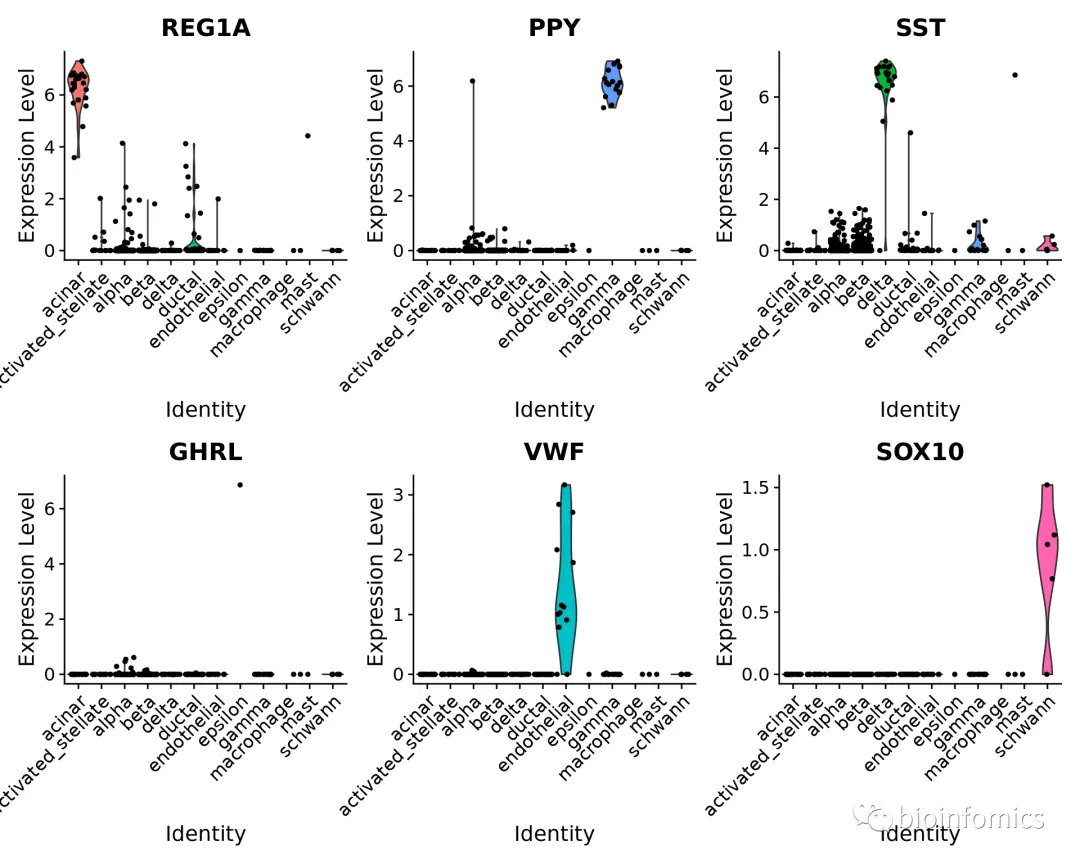
table(pancreas.query$predicted.id)#### acinar activated_stellate alpha beta## 21 17 248 258## delta ductal endothelial epsilon## 22 33 13 1## gamma macrophage mast schwann## 17 1 2 5VlnPlot(pancreas.query, c("REG1A", "PPY", "SST", "GHRL", "VWF", "SOX10"), group.by = "predicted.id")
二、使用SCTransform方法进行整合分析
Here, instead, we will harmonize the Pearson residuals that are output from SCTransform. As demonstrated below, the workflow consists of the following steps:
- Create a list of Seurat objects to integrate
- Perform SCTransform normalization separately for each dataset
- Run the PrepSCTIntegration function on the object list
- Integrate datasets, and proceed with joint analysis
首先,构建Seurat对象列表,并分别对每个对象运行SCTransform进行数据标准化:
library(Seurat)library(ggplot2)library(patchwork)options(future.globals.maxSize = 4000 * 1024^2)data("panc8")pancreas.list <- SplitObject(panc8, split.by = "tech")pancreas.list <- pancreas.list[c("celseq", "celseq2", "fluidigmc1", "smartseq2")]for (i in 1:length(pancreas.list)) {pancreas.list[[i]] <- SCTransform(pancreas.list[[i]], verbose = FALSE)}
接下来,选择用于数据整合的一些features,并运行PrepSCTIntegration
pancreas.features <- SelectIntegrationFeatures(object.list = pancreas.list, nfeatures = 3000)pancreas.list <- PrepSCTIntegration(object.list = pancreas.list, anchor.features = pancreas.features, verbose = FALSE)
然后使用FindIntegrationAnchors识别anchors,并运行IntegrateData进行数据集的整合,确保设置了normalization.method = ‘SCT’。
pancreas.anchors <- FindIntegrationAnchors(object.list = pancreas.list, normalization.method = "SCT", anchor.features = pancreas.features, verbose = FALSE)pancreas.integrated <- IntegrateData(anchorset = pancreas.anchors, normalization.method = "SCT", verbose = FALSE)
对整合后的数据进行下游的降维可视化
pancreas.integrated <- RunPCA(pancreas.integrated, verbose = FALSE)pancreas.integrated <- RunUMAP(pancreas.integrated, dims = 1:30)plots <- DimPlot(pancreas.integrated, group.by = c("tech", "celltype"))plots & theme(legend.position = "top") & guides(color = guide_legend(nrow = 3, byrow = TRUE, override.aes = list(size = 3)))
三、基于Reference-based的方法进行整合分析
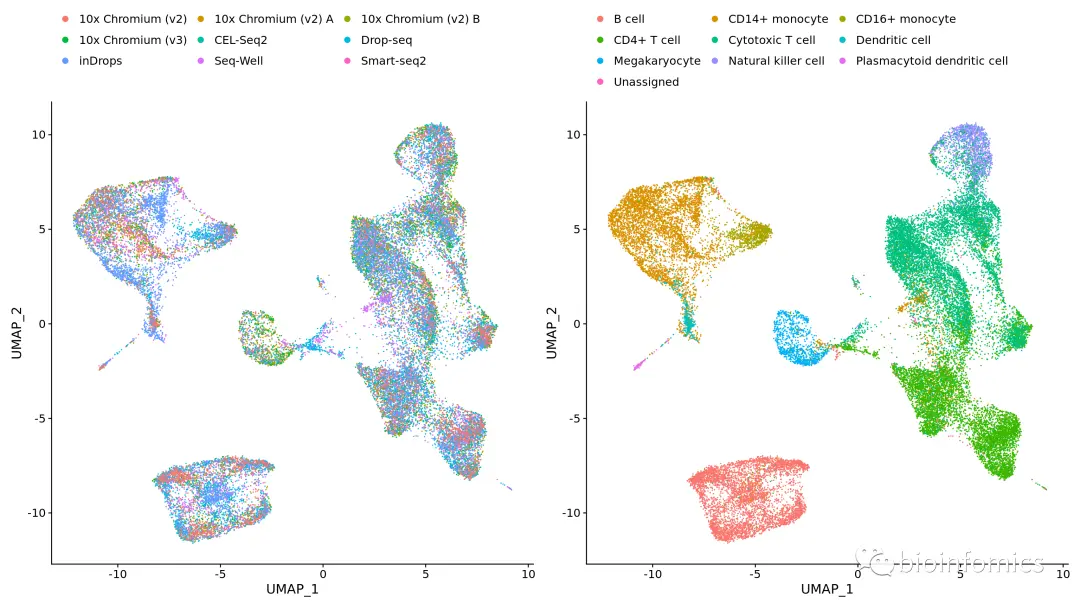
As an alternative, we introduce here the possibility of specifying one or more of the datasets as the ‘reference’ for integrated analysis, with the remainder designated as ‘query’ datasets. In this workflow, we do not identify anchors between pairs of query datasets, reducing the number of comparisons. For example, when integrating 10 datasets with one specified as a reference, we perform only 9 comparisons. Reference-based integration can be applied to either log-normalized or SCTransform-normalized datasets.
library(Seurat)library(SeuratData)library(ggplot2)library(patchwork)InstallData("pbmcsca")data("pbmcsca")# 分割数据集构建Seurat对象列表pbmc.list <- SplitObject(pbmcsca, split.by = "Method")# 分别对每个对象进行SCTransform标准化处理for (i in names(pbmc.list)) {pbmc.list[[i]] <- SCTransform(pbmc.list[[i]], verbose = FALSE)}# 选择用于数据集整合的featurespbmc.features <- SelectIntegrationFeatures(object.list = pbmc.list, nfeatures = 3000)# 执行PrepSCTIntegration处理pbmc.list <- PrepSCTIntegration(object.list = pbmc.list, anchor.features = pbmc.features)# 选择参考数据集reference_dataset <- which(names(pbmc.list) == "10x Chromium (v3)")# 识别整合的anchorspbmc.anchors <- FindIntegrationAnchors(object.list = pbmc.list, normalization.method = "SCT", anchor.features = pbmc.features, reference = reference_dataset)# 进行数据整合pbmc.integrated <- IntegrateData(anchorset = pbmc.anchors, normalization.method = "SCT")# 数据降维可视化pbmc.integrated <- RunPCA(object = pbmc.integrated, verbose = FALSE)pbmc.integrated <- RunUMAP(object = pbmc.integrated, dims = 1:30)plots <- DimPlot(pbmc.integrated, group.by = c("Method", "CellType"))plots & theme(legend.position = "top") & guides(color = guide_legend(nrow = 4, byrow = TRUE, override.aes = list(size = 2.5)))
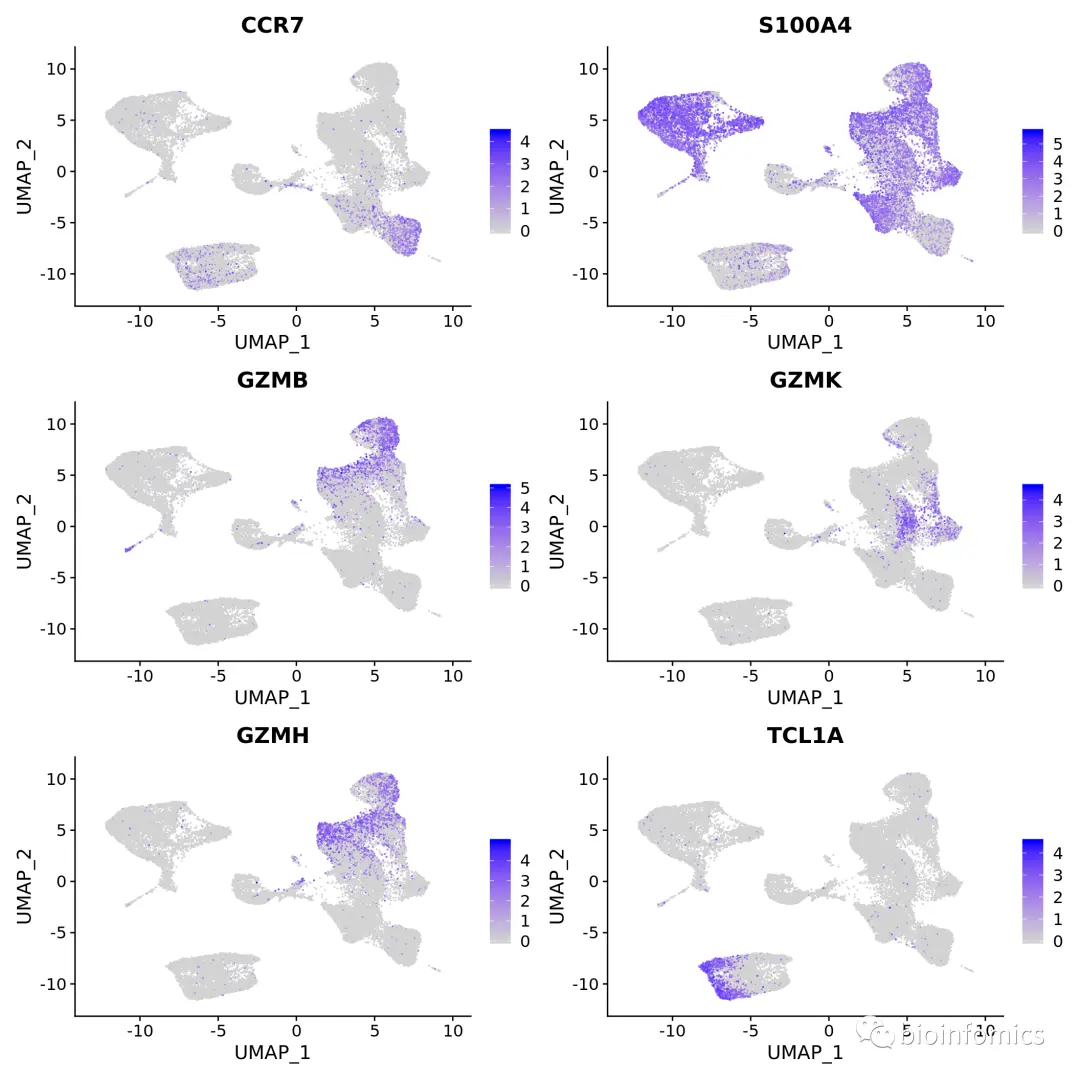
DefaultAssay(pbmc.integrated) <- "RNA"# Normalize RNA data for visualization purposespbmc.integrated <- NormalizeData(pbmc.integrated, verbose = FALSE)FeaturePlot(pbmc.integrated, c("CCR7", "S100A4", "GZMB", "GZMK", "GZMH", "TCL1A"))
sessionInfo()R version 3.6.0 (2019-04-26)Platform: x86_64-redhat-linux-gnu (64-bit)Running under: CentOS Linux 7 (Core)Matrix products: defaultBLAS/LAPACK: /usr/lib64/R/lib/libRblas.solocale:[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8[7] LC_PAPER=en_US.UTF-8 LC_NAME=C[9] LC_ADDRESS=C LC_TELEPHONE=C[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=Cattached base packages:[1] splines parallel stats4 grid stats graphics grDevices[8] utils datasets methods baseother attached packages:[1] loomR_0.2.1.9000 hdf5r_1.3.1[3] R6_2.4.0 mclust_5.4.5[5] garnett_0.1.16 org.Hs.eg.db_3.8.2[7] AnnotationDbi_1.46.0 pagedown_0.9.1[9] devtools_2.1.0 usethis_1.5.1[11] celaref_1.2.0 scran_1.12.1[13] scRNAseq_1.10.0 FLOWMAPR_1.2.0[15] Seurat_3.1.4.9902 sctransform_0.2.0[17] patchwork_0.0.1 cowplot_1.0.0[19] DT_0.12 RColorBrewer_1.1-2[21] shinydashboard_0.7.1 ggsci_2.9[23] shiny_1.3.2 stxBrain.SeuratData_0.1.1[25] pbmcsca.SeuratData_3.0.0 panc8.SeuratData_3.0.2[27] SeuratData_0.2.1 doParallel_1.0.14[29] iterators_1.0.12 binless_0.15.1[31] RcppEigen_0.3.3.5.0 foreach_1.4.7[33] scales_1.0.0 dplyr_0.8.3[35] pagoda2_0.1.0 harmony_1.0[37] Rcpp_1.0.2 conos_1.1.2

若有收获,就点个赞吧
0 人点赞
