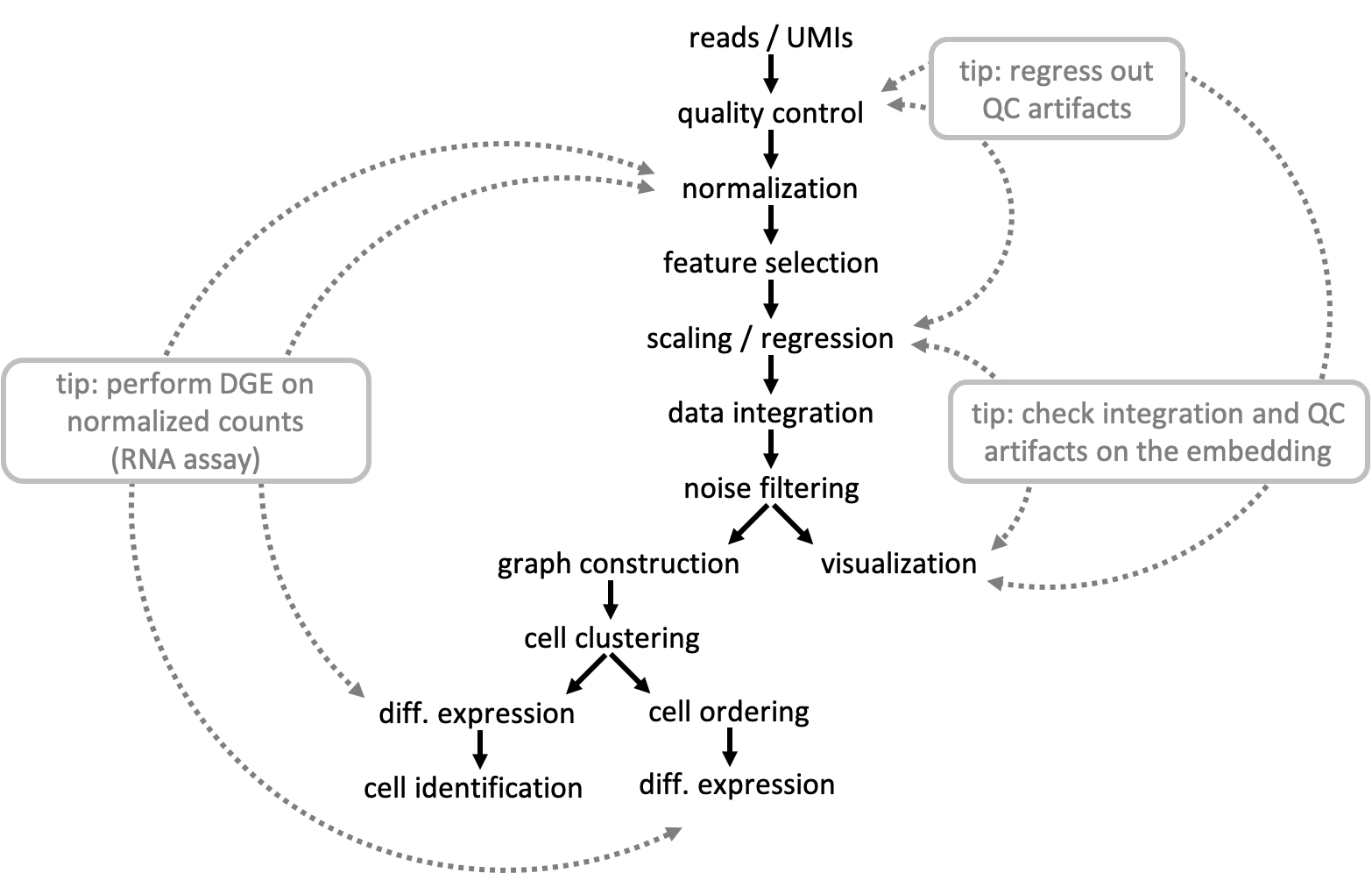
- Downloading data
- Seurat Objects
- Read .csv, .tsv, or .txt formats
- Convert to sparse matrix
- Read and convert from the .mtx format (it reads the files in the folder)
- Read and convert from the .h5 format
- Quality control
- Normalization and Regression
- Dimensionality reduction
- Creating graphs
- Dataset integration and batch correction
- Clustering
- Differential expression
- Trajectory inference
- Additional material
Downloading data
Running bash code in RStudio
It is possible to download data from within RStudio by using the bash code chunk. It is usually a standard to have a folder with all your raw data stored in a separate place from your code or analysis results.
RMarkdown also allows the use of languages other than R, such as bash or Python. To use bash, start the code chunk with {bash} instead of {r}:
# Make a data directorymkdir data# Use curl to download the datacurl -o data/FILENAME.h5 -O http://FILE_PATH.h5
Seurat Objects
Seurat is one of the most commonly used pipelines for scRNA-seq analysis. For additional information on Seurat objects and commands, please look at this page. Many different tutorials are also available from their website.
Reading files
There are many formats available in which one can store single cell information, many of which cannot all be listed here. The most common formats are:
- Using tab-delimited matrices saved as .csv, .tsv or .txt and with and additional matrix containing the sample metadata, which is common for SMARTseq2 and related methods
- Using a compressed sparse matrix file .mtx with annotations for genes and cells saved as .tsv, which was one of the defaults for 10X Chromium data
- Using HDF5 compressed file for in-file read-write access, which is now becoming the default method for storing single cell dataset (is the current default for 10X Chromium data). HDF5 in particular is fast, scalable and can load parts of the data that will be used at a time, and also can store the metadata in the same file, making it portable. It stores the data as binary compressed sparse matrix format
```r
Read .csv, .tsv, or .txt formats
raw_matrix <- read.delim( file = “data/folder_sample1.csv”, row.names = 1 )
Convert to sparse matrix
sparse_matrix <- Matrix::Matrix( data = raw_matrix, sparse = T) rm(raw_matrix)
Read and convert from the .mtx format (it reads the files in the folder)
sparse_matrix <- Seurat::Read10X( data.dir = “data/folder_sample1”)
Read and convert from the .h5 format
sparse_matrix <- Seurat::Read10X_h5( filename = “data/matrix_file.h5”, use.names = T)
Additionally, one should also import any associated metadata file as a data.frame, which opposed to a matrix can store both numbers, characters, booleans and factors.```r# From .csv or .tsv formatsmetadata <- read.delim(file = "data/metadata.csv",sep = ",",row.names = 1 )
Creating Seurat objects
In order to make the data analysis process a bit simpler, several single cell developers have implemented their own way of storing the data in a concise format in R and python (called objects).
We can now load the expression matrices into objects and then later merge them into a single merged object. Each analysis workflow (Seurat, Scater, Scranpy, etc) has its own way of storing data and here is how you can create a Seurat Object:
SeuratObject <- CreateSeuratObject(
counts = sparse_matrix,
assay = "RNA",
project = "SAMPLE1",
meta.data = metadata)
The CreateSeuratObject function can take as input a sparse matrix or a regular matrix with counts.
Understanding Seurat objects
Seurat objects have a easy way to access their contents using the @ or the $ characters after the object name: The _@_ _attribute allows you to access to all analysis slots including: `_assays_, _meta.data_, _graphs_ and _reduction_ slots. _ The $ sign allows you to access the columns of the metadata (just like you normally would do in a data.frame) in your Seurat object directly so thatSeuratObject$column1is equal toSeuratObject@meta.data$column1`.
By default, the data is loaded into an assay slot named RNA, but you can change the names of the slots when creating them (e.g., when creating the seurat object, or computing the reductions). Therefore, check the options in each of the Seurat functions to know where you are storing the data. Each assay contains information about the raw counts (counts), the normalized counts (data), the scaled/regressed data (scale.data) as well as information about the dispersion of genes (var). Additional assays will be created when doing data analysis, for example when performing data integration, you might store the data as new assay or as a new reduction slot (depending on the integration method used).
SeuratObject@ # Type this and press TAB in RStudio
SeuratObject@assays$ # Type this and press TAB in RStudio
SeuratObject@assays$RNA@ # Type this and press TAB in RStudio
One can check the dimensions and subset Seurat Objects as a dataframe using dim(SeuratObject).
For a very detailed explanation of all slots in the Seurat objects, please refer to the Seurat wiki.
Add in a metadata column
You can simply add a column by using the $ sign to allocate a vector to a metadata column.
SeuratObject$NEW_COLUMN_NAME <- setNames(
colnames(SeuratObject),
vector_NEW_DATA)
Or use the function **AddMetaData** to add one or multiple columns:
SeuratObject <- AddMetaData(
object = SeuratObject,
metadata = vector_NEW_DATA,
col.name = "NEW_COLUMN_NAME")
Subsetting Seurat objects
To subset a Seurat object, you can do it as if was a dataframe:
# Subsetting Features
SeuratObject <- SeuratObject[vector_FEATURES_TO_USE, ]
# Subsetting Cells
SeuratObject <- SeuratObject[, vector_CELLS_TO_USE ]
Plotting functions
The most common functions to use for plotting are the violin plot and the scatter plots for the dimensionality reduction calculated (you need to calculate it before using the function!).
To plot continuous variables as violin (press TAB inside the functions for more options):
VlnPlot(object = SeuratObject,
group.by = "orig.ident",
features = c("percent_mito"),
pt.size = 0.1,
ncol = 4,
y.max = 100) +
NoLegend()
To plot continuous variables as scatter plot using the umap reduction slot (press TAB inside the functions for more options):
FeaturePlot(object = SeuratObject,
features = c("FEATURE_1","FEATURE_2","FEATURE_3"),
reduction = "umap",
dims = c(1,2),
order = TRUE,
pt.size = .1,
ncol = 3)
To plot CATEGORICAL variables as scatter plot using the umap reduction slot (press TAB inside the functions for more options):
DimPlot(object = SeuratObject,
group.by = c("DATASET"),
reduction = "umap",
dims = c(1, 2),
pt.size = .1,
label = TRUE,
ncol = 3)
Many other plotting functions are available, check Seurat:: (then press tab and look for the functions with “Plot” in the name).
Combine datasets
CombinedSeuratObject <- merge(
x = SeuratObject1,
y = c(SeuratObject2, SeuratObject3, SeuratObject4),
add.cell.ids = c("Dataset1", "Dataset2", "Dataset3", "Dataset4"))
Quality control
A very crucial step in scRNA-seq analysis is Quality control (QC). There will always be some failed libraries, low quality cells and doublets in an scRNA-seq dataset, hence the quality of the cells need to be examined and possibly some cells need to be removed.
Total number of features
A standard approach is to filter cells with low amount of reads as well as genes that are present in at least a certain amount of cells. Here we will only consider cells with at least 200 detected genes and genes need to be expressed in at least 3 cells. Please note that those values are highly dependent on the library preparation method used. Extremely high number of detected genes could indicate doublets. However, depending on the cell type composition in your sample, you may have cells with higher number of genes (and also higher counts) from one cell type.
VlnPlot(SeuratObject,
group.by = "orig.ident",
features = c("nFeature_RNA", "nCount_RNA"),
pt.size = 0.1,
ncol = 4) +
NoLegend()
Gene QC
In single cell, the most detected genes usually belong to housekeeping gene families, such as mitochondrial (MT-), ribosomal (RPL and RPS) and other structural proteins (i.e., ACTB, TMSB4X, B2M, EEF1A1).
# Compute the relative expression of each gene per cell
rel_expression <-
Matrix::t(Matrix::t(SeuratObject@assays$RNA@counts) /
Matrix::colSums(SeuratObject@assays$RNA@counts)) * 100
most_expressed <-
sort(Matrix::rowSums(rel_expression), TRUE) / ncol(SeuratObject)
# Plot the relative expression of each gene per cell
par(mfrow = c(1, 3),
mar = c(4, 6, 1, 1))
boxplot(as.matrix(Matrix::t(rel_expression[names(most_expressed[30:1]), ])),
cex = .1,
las = 1,
xlab = "% total count per cell",
col = scales::hue_pal()(90)[30:1],
horizontal = TRUE,
ylim = c(0, 8))
boxplot(as.matrix(Matrix::t(rel_expression[names(most_expressed[60:31]), ])),
cex = .1,
las = 1,
xlab = "% total count per cell",
col = scales::hue_pal()(90)[60:31],
horizontal = TRUE,
ylim = c(0, 8))
boxplot(as.matrix(Matrix::t(rel_expression[names(most_expressed[90:61]), ])),
cex = .1,
las = 1,
xlab = "% total count per cell",
col = scales::hue_pal()(90)[90:61],
horizontal = TRUE,
ylim = c(0, 8))
You might see that some genes constitute up to 10-30% of the counts from a single cell and the other top genes are mitochondrial and ribosomal genes. It is quite common that nuclear lincRNAs have correlation with quality and mitochondrial reads. Let us assemble some information about such genes, which are important for quality control and downstream filtering.
These genes can serve several purposes in single-cell data analysis, such as computing cell quality metrics (see below), normalize data (see below) and even help account for batch effects (Lin et al (2019) PNAS).
% Mitochondrial genes
Having the data in a suitable format, we can start calculating some quality metrics. We can for example calculate the percentage of mitocondrial and ribosomal genes per cell and add to the metadata. This will be helpfull to visualize them across different metadata parameteres (i.e. datasetID and chemistry version). There are several ways of doing this. Here is an example of how to manually calculate the proportion of mitochondrial reads and add to the metadata table.
Citing from “Simple Single Cell” workflows (Lun, McCarthy & Marioni, 2017): “High proportions are indicative of poor-quality cells (Islam et al. 2014; Ilicic et al. 2016), possibly because of loss of cytoplasmic RNA from perforated cells. The reasoning is that mitochondria are larger than individual transcript molecules and less likely to escape through tears in the cell membrane.”
(PS: non-linear relationship)
# Calculating % mitochondrial genes
SeuratObject <- PercentageFeatureSet(
object = SeuratObject,
pattern = "^RP[SL]",
assay = "RNA",
col.name = "percent_mito")
Note that you might need to change the patterns if you are using an organism other than human, e.g. “^Mt-“ for mouse.
% Ribosomal genes
In the same manner we will calculate the proportion gene expression that comes from ribosomal proteins. Ribosomal genes are the also among the top expressed genes in any cell and, on the contrary to mitochondrial genes, are inversely proportional to the mitochondrial content: the higher the mitochondrial content, the lower is the detection of ribosomal genes (PS: non-linear relationship).
# Calculating % ribosomal genes
SeuratObject <- PercentageFeatureSet(
SeuratObject,
pattern = "^RP[SL]",
col.name = "percent_ribo")
% Gene biotype and chromosome location
In RNA-sequencing, genes can be categorized into different groups depending on their RNA biotype. For example, “coding”, “non-coding”, “VDJ region genes” are “small interfering RNA” common gene biotypes. Besides, having information about chromosomal location might be useful to identify batch effects driven by sex chromosomes
Depending on the desired type of analysis, some gene categories can be filtered out if not of interest. For single cell specifically, cell libraries are usually constructed using poly-A enrichment and therefore enriching for “protein-coding proteins”, which usually constitutes around 80-90% of all available genes.
# Retrieve mouse gene annotation from ENSEMBL
library(biomaRt)
mart = biomaRt::useMart(
biomart = "ensembl",
dataset = "mmusculus_gene_ensembl",
host = "apr2020.archive.ensembl.org")
# Retrieve the selected attributes mouse gene annotation
annot <- biomaRt::getBM(
mart = mart,
attributes = c("external_gene_name", "gene_biotype", "chromosome_name"))
Make sure you are using the right species for your dataset. A list of all species available on can be found using biomaRt::listDatasets(mart)[,"dataset"]. All species names are formatted in the same way, such as mmusculus_gene_ensembl and hsapiens_gene_ensembl. For reproducibility reasons, it is also advised to specifically choose a biomart release version, since some genes will be renamed, inserted or deleted from the database. You can do so by running biomaRt::listEnsemblArchives().
# Match the gene names with theit respective gene biotype
item <- annot[match(rownames(SeuratObject@assays$RNA@counts), annot[, 1]), "gene_biotype"]
item[is.na(item)] <- "unknown"
# Calculate the percentage of each gene biotype
perc <- rowsum(as.matrix(SeuratObject@assays$RNA@counts), group = item)
perc <- (t(perc) / Matrix::colSums(SeuratObject@assays$RNA@counts))
o <- order(apply(perc, 2, median), decreasing = FALSE)
perc <- perc[, o]
# Plot percentage of each gene biotype
boxplot(perc * 100,
outline = FALSE,
las = 2,
main = "% reads per cell",
col = scales::hue_pal()(100),
horizontal = TRUE)
# Add table to the object
gene_biotype_table <-
setNames(as.data.frame((perc*100)[, names(sort(table(item), decreasing = TRUE))]),
paste0("percent_", names(sort(table(item), decreasing = TRUE))))
SeuratObject@meta.data <-
SeuratObject@meta.data[, !(colnames(SeuratObject@meta.data) %in% colnames(gene_biotype_table))]
SeuratObject@meta.data <- cbind(
SeuratObject@meta.data,
gene_biotype_table)
The code above can also be done again by replacing the string "gene_biotype" by "chromosome_name":
# Match the gene names with their respective chromosome location
item <- annot[match(rownames(SeuratObject@assays$RNA@counts), annot[, 1]),
"chromosome_name"]
item[is.na(item)] <- "unknown"
item[!item %in% as.character(c(1:23, "X", "Y", "MT"))] <- "other"
If you want to focus the analysis on only protein-coding genes, for example, you can do it like so:
dim(SeuratObject)
sel <- annot[match(rownames(SeuratObject), annot[, 1]), 2] == "protein_coding"
genes_use <- rownames(SeuratObject)[sel]
genes_use <- as.character(na.omit(genes_use))
SeuratObject <- SeuratObject[genes_use, ]
dim(SeuratObject)
Cell cycle scoring
We here perform cell cycle scoring. To score a gene list, the algorithm calculates the difference of mean expression of the given list and the mean expression of reference genes. To build the reference, the function randomly chooses a bunch of genes matching the distribution of the expression of the given list. Cell cycle scoring with Seurat adds three slots in data, a score for S phase, a score for G2M phase and the predicted cell cycle phase. The Seurat package provides a list of human G2M and S phase genes in cc.genes.
How to run it:
SeuratObject <- CellCycleScoring(
object = SeuratObject,
g2m.features = cc.genes$g2m.genes,
s.features = cc.genes$s.genes)
SeuratObject$G1.Score <- 1 - SeuratObject$S.Score - SeuratObject$G2M.Score
Visualizing all metadata in a PCA plot
Having many metadata parameters to analyse individually makes it a bit hard to visualize the real differences between datasets, batches and experimental conditions. One way to try to combine all this information into one plot is by running dimensionality reduction via principal component analysis (PCA) on the continuous variables in the metadata. Thus visualizing on the top principal components (1st and 2nd) reflects how different the datasets are.
# Calculate PCA using selected metadata parameters
metadata_use <- grep("perc", colnames(SeuratObject@meta.data), value = TRUE)
metadata_use <- c("nCount_RNA", "nFeature_RNA", "S.Score", "G2M.Score", metadata_use)
# Remove columns in the metadata that are all 0s
metadata_use <- metadata_use[colSums(SeuratObject@meta.data[, metadata_use] != 0 ) > 0 ]
# Run PCA
PC <- prcomp(SeuratObject@meta.data[, metadata_use],
center = TRUE,
scale. = TRUE)
# Add the PCA (ran on the METADATA) in the object
SeuratObject@reductions[["pca_metadata"]] <- CreateDimReducObject(
embeddings = PC$x,
key = "metadataPC_",
assay = "RNA")
# Plot the PCA ran on the METADATA
DimPlot(SeuratObject,
reduction = "pca_metadata",
dims = c(1, 2),
group.by = "orig.ident" )
N.B. Zero-count genes and identical/all-zero metadata columns (i.e. any value that does that have any variation) will interfere with analyses such as PCA. You will thus have to either remove those zero-count genes and/or exclude some metadata columns:
# Remove zero-count genes
SeuratObject <- SeuratObject[Matrix::rowSums(SeuratObject) > 0, ]
# Exclude metadata columns that are zero
metadata_use <- metadata_use[colSums(SeuratObject@meta.data[, metadata_use] != 0 ) > 0 ]
Normalization and Regression
Before doing any other analyses, the data needs to be normalized to account for varying sequencing depths and logtransformed (in most cases). Furthermore, it may be useful to regress out confounding factors, for example cell cycle or quality metrics, such as percent mitochondria or number of detected genes.
Normalization
The most common normalization for RNA-seq and also single-cell RNA-seq is log-normalization. This is done by dividing the gene counts of each gene by the sum of all gene counts (a.k.a., library size) to compensate for library size differences. Then the result is multiplied by a constant number, so all cell have the same sequencing depth. For bulk RNA-seq, the constant is usually 1e6, resulting in CPM (counts per million), but since single-cells library sizes are way lower than that, the number ranges from 1e3 to 1e4 (counts per 10000).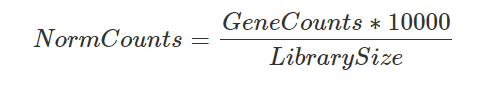
The library size-corrected values are then log-transformed to achieve a log-normal data distribution.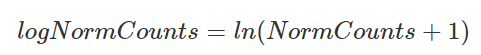
# Remove genes with zero variance
SeuratObject <- SeuratObject[ Matrix::rowSums(SeuratObject) > 0, ]
# Remove genes with zero variance
SeuratObject <- NormalizeData(
object = SeuratObject,
scale.factor = 10000,
normalization.method = "LogNormalize")
Feature selection
An important step in many big-data analysis tasks is to identify features (genes, transcripts, proteins, metabolites, etc) that are actually very variable between the samples being looked at.
For example. Imagine that you have a dataset known to contain different types of epithelial cells, and you use either 1) only genes that are expressed and shared across all epithelial cells at about the same level, 2) only genes that are not detected in epithelial cells, 3) only genes which expression differ greatly across epithelial cells or 4) using all genes. Which of these 4 gene lists can best distinguish the epithelial subtypes in this dataset?
As you could now imagine, using only genes which expression differ greatly across epithelial cells is the best case scenario, followed by using all genes. Therefore, using only genes that are expressed and shared across all epithelial cells at about the same level or only genes that are not detected in epithelial cells do not contain sufficient information to distinguish the epithelial subtypes.
However, since in single-cell we usually do not know the epithelial subtypes the cells before hand (since this is what we want to discover), we need another method to accomplish this task. In general terms, a common approach is to order genes by their overall variance across samples. This is because genes with higher variance will also likely be the ones that can separate the cells the best.
Since genes with higher expression level usually also have naturally higher variation, the gene variation is then normalized by the log mean expression of each gene (see plot).
# Find the variable features
SeuratObject <- FindVariableFeatures(
object = SeuratObject,
nfeatures = 3000,
selection.method = "vst",
verbose = FALSE,
assay = "RNA")
# Plot the top 20 variable features
top20 <- head(VariableFeatures(SeuratObject), 20)
LabelPoints(plot = VariableFeaturePlot(SeuratObject),
points = top20,
repel = TRUE)
Scaling and Centering (linear)
Since each gene has a different expression level, it means that genes with higher expression values will naturally have higher variation that will be captured by downstream methods. This means that we need to somehow give each gene a similar weight beforehand (see below). A common practice is to center and scale each gene before performing PCA. This exact scaling is called Z-score normalization it is very useful for PCA, clustering and plotting heatmaps.
Additionally, we can use regression to remove any unwanted sources of variation from the dataset, such as cell cycle, sequencing depth, percent mitochondria. This is achieved by doing a generalized linear regression (GLM) using these parameters as covariates in the model. Then the residuals of the model are taken as the “regressed data”. Although perhaps not in the best way, batch effect regression can also be done here.
SeuratObject <- ScaleData(
object = SeuratObject,
vars.to.regress = c("nCount_RNA", "mito.percent", "nFeatures_RNA"),
model.use = "linear",
assay = "RNA",
do.scale = TRUE,
do.center = TRUE)
Seurat will be default run scaling on any variable features in a Seurat object, if they exists. This can greatly speed up the performance of scaling step while the results of downstream analyses (such as dimensionality reduction or clustering) remain unchanged, as those are based on the variable genes themselves. If you’d rather scale the data on the full number of genes even after you’ve used FindVariableFeatures, you can specify this in the function call: ScaleData(..., features = rownames(SeuratObject)).
Scaling and Centering (poisson)
Since the procedure above assumes a log-linear data distribution, it may be the case that it does not regress the variation correctly, as RNA-seq data (including single cell) relates more closely to a negative binomial distribution. An alternative variation of the procedure above can also be run on the raw UMI count data but using a “poisson” or “negative binomial” distribution instead. This is performing a gene-wise GLM regression using a poisson model.
SeuratObject <- ScaleData(
object = SeuratObject,
vars.to.regress = c("nCount_RNA", "mito.percent", "nFeatures_RNA"),
model.use = "poisson",
assay = "RNA",
do.scale = TRUE,
do.center = TRUE)
SCtransform
Scaling and centering assuming a poisson distribution might in some cases overfit the data, see above. One can overcome this by pooling information across genes with similar abundances in order to obtain more stable parameter estimates to be used as gene weights in the regression model. This is called “scTransform” and, in simple terms, is performing a gene-wise GLM regression using a constrained negative binomial model.
SeuratObject <- SCTransform(
object = SeuratObject,
assay = "RNA",
vars.to.regress = c("nCount_RNA", "mito.percent", "nFeatures_RNA"),
new.assay.name = "sctransform",
do.center = TRUE)
Dimensionality reduction
The full gene expression space, with thousands of genes, contains quite a lot of noise in scRNA-seq data and is hard to visualize. Hence, most scRNA-seq analyses starts with a step of PCA (or similar method, e.g. ICA) to remove some of the variation of the data.
For a simple scRNA-seq dataset with only a few cell types, PCA may be sufficient to visualize the complexity of the data in 2 or 3 dimensions. However, with increasing complexity we need to run non-linear dimensionality reduction to be able to project the data down to 2 dimensions for visualization, such methods are tSNE, UMAP and diffusion maps.
Deciding which dimensionality reduction method to use is a non-trivial question, one that may have several good answers. Here is a paper comparing many of the methods available, if you want to read more details.
PCA
Principal Component Analysis (PCA) is defined as an orthogonal linear transformation that transforms the data to a new coordinate system such that the greatest variance by some scalar projection of the data comes to lie on the first coordinate (called the first principal component), the second greatest variance on the second coordinate, and so on. […] Often, its operation can be thought of as revealing the internal structure of the data in a way that best explains the variance in the data. […] This is done by using only the first few principal components so that the dimensionality of the transformed data is reduced.
Adapted from Wikipedia
# Run pca
SeuratObject <- RunPCA(
object = SeuratObject,
assay = "RNA",
npcs = 100,
verbose = FALSE )
tSNE
Maaten, Hilton (2008) J of Machine Learning Research
t-distributed stochastic neighborhood embedding (tSNE) is a nonlinear dimensionality reduction technique well-suited for embedding high-dimensional data for visualization in a low-dimensional space of two or three dimensions. Specifically, it models each high-dimensional object by a two- or three-dimensional point in such a way that similar objects are modeled by nearby points and dissimilar objects are modeled by distant points with high probability. t-SNE has been used for visualization in a wide range of applications, including […] bioinformatics […]. While t-SNE plots often seem to display clusters, the visual clusters can be influenced strongly by the chosen parameterization and therefore a good understanding of the parameters for t-SNE is necessary.
Adapted from Wikipedia
Useful links:
How to Use t-SNE Effectively
SeuratObject <- RunTSNE(object = SeuratObject,
reduction = "pca",
perplexity = 30,
max_iter = 1000,
theta = 0.5,
eta = 200,
exaggeration_factor = 12,
dims.use = 1:50,
verbose = TRUE,
num_threads = 0)
UMAP
Uniform Manifold Approximation and Projection (UMAP) is a dimension reduction technique that can be used for visualization similarly to t-SNE, but also for general non-linear dimension reduction.
umap-learn documentation
The result is a practical scalable algorithm that applies to real world data. The UMAP algorithm is competitive with t-SNE for visualization quality, and arguably preserves more of the global structure with superior run time performance. Furthermore, UMAP has no computational restrictions on embedding dimension, making it viable as a general purpose dimension reduction technique for machine learning.
UMAP Arxiv paper
SeuratObject <- RunUMAP(object = SeuratObject,
reduction = "pca",
dims = 1:top_PCs,
n.components = 2,
n.neighbors = 20,
spread = .3,
repulsion.strength = 1,
min.dist = .001,
verbose = TRUE,
num_threads = 0,
n.epochs = 200,
metric = "euclidean",
seed.use = 42,
reduction.name = "umap")
DM
Diffusion maps (DM) is a dimensionality reduction […] which computes a family of embeddings of a data set into Euclidean space (often low-dimensional) whose coordinates can be computed from the eigenvectors and eigenvalues of a diffusion operator on the data. The Euclidean distance between points in the embedded space is equal to the “diffusion distance” between probability distributions centered at those points. Different from linear dimensionality reduction methods such as principal component analysis (PCA) and multi-dimensional scaling (MDS), diffusion maps is part of the family of nonlinear dimensionality reduction methods which focus on discovering the underlying manifold that the data has been sampled from. […] The basic observation is that if we take a random walk on the data, walking to a nearby data-point is more likely than walking to another that is far away.
Wikipedia
Diffusion Maps paper
# Load additional libraries
library(destiny)
# Run diffusion maps using the destiny package
dm <- DiffusionMap(data = SeuratObject@reductions[["pca"]]@cell.embeddings[, 1:50],
k = 20,
n_eigs = 20)
# Fix the cell names in the DM embedding
rownames(dm@eigenvectors) <- colnames(SeuratObject)
# Add the DM embbedding to the SeuratObject
SeuratObject@reductions[["dm"]] <- CreateDimReducObject(embeddings = dm@eigenvectors,
key = "DC_",
assay = "RNA")
ICA
Independent Component Analysis (ICA) is a computational method for separating a multivariate signal into additive subcomponents. This is done by assuming that the subcomponents are non-Gaussian signals and that they are statistically independent from each other. ICA is a special case of blind source separation.
Wikipedia
SeuratObject <- RunICA(object = SeuratObject,
assay = "pca",
nics = 20,
reduction.name = "ica")
Creating graphs
Instead of doing clustering of scRNA-seq data on the full expression matrix or in PCA space (which gives linear distances), it has proven quite powerful to use graphs to create a non-linear representation of cell-to-cell similarities.
Graphs is simply a representation of all cells (as nodes/vertices) with edges drawn between them based on some similarity criteria. For instance, a graph can be constructed with edges between all cells that are less than X distance from each other in PCA space with 50 principal components.
KNN
KNN refers to “K Nearest Neighbors”, which is a basic and popular topic in data mining and machine learning areas. The KNN graph is a graph in which two vertices p and q are connected by an edge, if the distance between p and q is among the K-th smallest distances.[2] Given different similarity measure of these vectors, the pairwise distance can be Hamming distance, Cosine distance, Euclidean distance and so on. We take Euclidean distance as the way to measure similarity between vectors in this paper. The KNN Graph data structure has many advantages in data mining. For example, for a billion-level dataset, pre-building a KNN graph offline as an index is much better than doing KNN search online many times.
Adapted from Github
SeuratObject <- FindNeighbors(SeuratObject,
assay = "RNA",
compute.SNN = FALSE,
reduction = "pca",
dims = 1:50,
graph.name = "SNN",
prune.SNN = 1/15,
k.param = 20,
force.recalc = TRUE)
Setting compute.SNN toFALSE will only compute the KNN graph.
We can take a look at the KNN graph. It is a matrix where every connection between cells is represented as 1s. This is called a unweighted graph (default in Seurat). Some cell connections can however have more importance than others, in that case the scale of the graph from 0 to a maximum distance. Usually, the smaller the distance, the closer two points are, and stronger is their connection. This is called a weighted graph. Both weighted and unweighted graphs are suitable for clustering, but clustering on unweighted graphs is faster for large datasets (> 100k cells).
library(pheatmap)
pheatmap(SeuratObject@graphs$CCA_nn[1:200, 1:200],
col = c("white", "black"),
border_color = "grey90",
legend = FALSE,
cluster_rows = FALSE,
cluster_cols = FALSE,
fontsize = 2)
SNN
In addition to the KNN graph, if we then determine the number of nearest neighbors shared by any two points. In graph terminology, we form what we call the “shared nearest neighbor” graph. We do this by replacing the weight of each link between two points (in the nearest neighbor graph) by the number of neighbors that the points share. In other words, this is the number of length 2 paths between any two points in the nearest neighbor graph.
After, this shared nearest neighbor graph is created, all pairs of points are compared and if any two points share more than T neighbors, i.e., have a link in the shared nearest neighbor graph with a weight more than our threshold value, T( TS:. n), then the two points and any cluster they are part of are merged. In other words, clusters are connected components in our shared nearest neighbor graph after we sparsify using a threshold.
SeuratObject <- FindNeighbors(SeuratObject,
assay = "RNA",
compute.SNN = T,
reduction = "pca" ,
dims = 1:50,
graph.name = "SNN",
prune.SNN = 1/15,
k.param = 20,
force.recalc = TRUE)
Settingcompute.SNN to TRUE will compute both the KNN and SNN graphs.
Dataset integration and batch correction
Existing batch correction methods were specifically designed for bulk RNA-seq. Thus, their applications to scRNA-seq data assume that the composition of the cell population within each batch is identical. Any systematic differences in the mean gene expression between batches are attributed to technical differences that can be regressed out. However, in practice, population composition is usually not identical across batches in scRNA-seq studies. Even assuming that the same cell types are present in each batch, the abundance of each cell type in the data set can change depending upon subtle differences in cell culture or tissue extraction, dissociation and sorting, etc. Consequently, the estimated coefficients for the batch blocking factors are not purely technical, but contain a non-zero biological component due to differences in composition. Batch correction based on these coefficients will thus yield inaccurate representations of the cellular expression proles, potentially yielding worse results than if no correction was performed.
Haghverdi et al (2018) Nat Biotechnology
As with many of the other scRNA-seq methodologies, it can be difficult to choose which batch-correction technique you should use for your dataset. You can read more in this comparative paper.
MNN
An alternative approach for data merging and comparison in the presence of batch effects uses a set of landmarks from a reference data set to project new data onto the reference. The rationale here is that a given cell type in the reference batch is most similar to cells of its own type in the new batch. This strategy depends on the selection of landmark points in high dimensional space picked from the reference data set, which cover all cell types that might appear in the later batches. However, if the new batches include cell types that fall outside the transcriptional space explored in the reference batch, these cell types will not be projected to an appropriate position in the space defined by the landmarks. […] The difference in expression values between cells in a MNN pair provides an estimate of the batch effect, which is made more precise by averaging across many such pairs. A correction vector is obtained from the estimated batch effect and applied to the expression values to perform batch correction. Our approach automatically identifies overlaps in population composition between batches and uses only the overlapping subsets for correction, thus avoiding the assumption of equal composition required by other methods.
The use of MNN pairs involves three assumptions: (i) there is at least one cell population that is present in both batches, (ii) the batch effect is almost orthogonal to the biological subspace, and (iii) batch effect variation is much smaller than the biological effect variation between different cell types.
Haghverdi et al (2018) Nat Biotechnology
# Load additional libraries
library(SeuratWrappers)
# Run MNN
SeuratObject.list <- SplitObject(SeuratObject, split.by = "BATCH")
SeuratObject <- RunFastMNN(object.list = SeuratObject.list,
assay = "RNA",
features = 2000,
reduction.name = "mnn")
# Free memory from working environment
rm(c(SeuratObject.list))
gc(verbose = FALSE)
CCA Anchors
Since MNNs have previously been identified using L2-normalized gene expression, significant differences across batches can obscure the accurate identification of MNNs, particularly when the batch effect is on a similar scale to the biological differences between cell states. To overcome this, we first jointly reduce the dimensionality of both datasets using diagonalized CCA, then apply L2-normalization to the canonical correlation vectors. We next search for MNNs in this shared low-dimensional representation. We refer to the resulting cell pairs as anchors, as they encode the cellular relationships across datasets that will form the basis for all subsequent integration analyses.
Obtaining an accurate set of anchors is paramount to successful integration. Aberrant anchors that form between different biological cell states across datasets are analogous to noisy edges that occur in k-nearest neighbor (KNN) graphs (Bendall et al., 2014) and can confound downstream analyses. This has motivated the use of shared nearest neighbor (SNN) graphs (Levine et al., 2015; Shekhar et al., 2016), where the similarity between two cells is assessed by the overlap in their local neighborhoods. As this measure effectively pools neighbor information across many cells, the result is robust to aberrant connections in the neighbor graph. We introduced an analogous procedure for the scoring of anchors, where each anchor pair was assigned a score based on the shared overlap of mutual neighborhoods for the two cells in a pair. High-scoring correspondences therefore represent cases where many similar cells in one dataset are predicted to correspond to the same group of similar cells in a second data- set, reflecting increased robustness in the association between the anchor cells. While we initially identify anchors in low-dimensional space, we also filter out anchors whose correspondence is not supported based on the original untransformed data.
Stuart et al (2019) Cell30559-8)
SeuratObject.list <- SplitObject(
object = SeuratObject,
split.by = "BATCH")
SeuratObject.anchors <- FindIntegrationAnchors(
object.list = SeuratObject.list,
dims = 1:30)
SeuratObject <- IntegrateData(
anchorset = SeuratObject.anchors,
dims = 1:30,
new.assay.name = "cca")
Scanorama
Scanorama’s approach generalizes mutual nearest-neighbors matching, a technique that finds similar elements between two datasets, to instead find similar elements among many datasets.
While recent approaches have shown that it is possible to integrate scRNA-seq studies across multiple experiments, these approaches automatically assume that all datasets share at least one cell type in common or that the gene expression profiles share largely the same correlation structure across all datasets. These methods are therefore prone to overcorrection, especially when integrating collections of datasets with considerable differences in cellular composition.
Scanorama is a strategy for efficiently integrating multiple scRNA-seq datasets, even when they are composed of heterogeneous transcriptional phenotypes. Its approach is analogous to computer vision algorithms for panorama stitching that identify images with overlapping content and merge these into a larger panorama. Likewise, Scanorama automatically identifies scRNA-seq datasets containing cells with similar transcriptional profiles and can leverage those matches for batch correction and integration, without also merging datasets that do not overlap. Scanorama is robust to different dataset sizes and sources, preserves dataset-specific populations and does not require that all datasets share at least one cell population.
edited from Welch et al (2019) Cell
# Split the object by batch
SeuratObject.list <- SplitObject(
object = SeuratObject,
split.by = "BATCH")
assaylist <- list()
genelist <- list()
for(i in 1:length(SeuratObject.list)) {
assaylist[[i]] <- t(as.matrix(GetAssayData(SeuratObject.list[[i]], "data")))
genelist[[i]] <- rownames(SeuratObject.list[[i]])
}
library(reticulate)
scanorama <- import("scanorama")
integrated.data <- scanorama$integrate(assaylist, genelist)
corrected.data <- scanorama$correct(assaylist,
genelist,
return_dense = TRUE)
integrated.corrected.data <- scanorama$correct(assaylist,
genelist,
return_dimred = TRUE,
return_dense = TRUE)
intdata <- lapply(integrated.corrected.data[[2]], t)
panorama <- do.call(cbind, intdata)
rownames(panorama) <- as.character(integrated.corrected.data[[3]])
colnames(panorama) <- unlist(sapply(assaylist, rownames))
intdimred <- do.call(rbind, integrated.corrected.data[[1]])
colnames(intdimred) <- paste0("PC_", 1:100)
# Add standard deviations in order to draw Elbow Plots in Seurat
stdevs <- apply(intdimred, MARGIN = 2, FUN = sd)
SeuratObject@assays[["pano"]] <- CreateAssayObject(data = panorama,
min.cells = 0,
min.features = 0)
SeuratObject[["pca_scanorama"]] <- CreateDimReducObject(
embeddings = intdimred,
stdev = stdevs,
key = "PC_",
assay = "pano")
Harmony
Harmony begins with a low-dimensional embedding of cells, such as principal components analysis (PCA), (Supplementary Note 1 and Methods). Using this embedding, Harmony first groups cells into multi-dataset clusters. We use soft clustering to assign cells to potentially multiple clusters, to account for smooth transitions between cell states. These clusters serve as surrogate variables, rather than to identify discrete cell types
After clustering, each dataset has cluster-specific centroids that are used to compute cluster-specific linear correction factors. Since clusters correspond to cell types and states, cluster-specific correction factors correspond to individual cell-type and cell-state specific correction factors. In this way, Harmony learns a simple linear adjustment function that is sensitive to intrinsic cellular phenotypes. Finally, each cell is assigned a cluster-weighted average of these terms and corrected by its cell-specific linear factor. Since each cell may be in multiple clusters, each cell has a potentially unique correction factor. Harmony iterates these four steps until cell cluster assignments are stable.
Harmony is an algorithm for robust, scalable and flexible multi-dataset integration to meet four key challenges of unsupervised scRNA-seq joint embedding: scaling to large datasets, identification of both broad populations and fine-grained subpopulations, flexibility to accommodate complex experimental design, and the power to integrate across modalities.
adapted from Korsunsky et al (2019) Nat Mathods
# Load additional libraries
library(harmony)
library(SeuratWrappers)
SeuratObject <- RunHarmony(
SeuratObject,
group.by.vars = "Method")
Clustering
One of the main goals of scRNA-seq studies is often to group cells into clusters of similar cells. There are many different ways of defining clusters. There are traditional clustering approaches like K-mean, hierarchical clustering and affinity propagation, but recently most pipelines make use of graphs (see section above) to define clusters as groups of interconnected cells.
To define clusters in a graph we make use of different community detection algorithms like Louvain, Leiden and Infomap. The main idea behind all of them is to find groups of cells that have more edges (connections of high similarity) between them than they have to other cells in the dataset.
Still, all clustering methods begin with defining pairwise distances between cells. Commonly this is done by taking euclidean distances between cells in PCA space, after selecting a subset of variables and scaling the data. Hence, your clustering results will be strongly dependent on choices you made in preprocessing of the data, especially the variable gene selection and how many principal components you include from the PCA.
In graph based methods, the distances from PCA space are used to create a graph with edges between neighboring cells. Also here, there are multiple parameters, like number of neighbors in the KNN graph and pruning settings for the SNN construction that will impact the clustering results. If you want to read more about clustering and comparisons between methods, have a look at this paper.
Louvain
The Louvain method for community detection is a method to extract communities from large networks created by Blondel et al. from the University of Louvain. The method is a greedy optimization method that appears to run in time O(n.log2n) in the number of nodes in the network.The value to be optimized is modularity, defined as a value in the range that measures the density of links inside communities compared to links between communities. Optimizing this value theoretically results in the best possible grouping of the nodes of a given network, however going through all possible iterations of the nodes into groups is impractical so heuristic algorithms are used.
Wikipedia
Louvain Paper
SeuratObject <- FindClusters(
object = SeuratObject,
graph.name = "SNN",
resolution = 0.8,
algorithm = 1) # Setting `algorithm = 1` signifies Louvain
The number of clusters can be controlled using theresolution parameter with higher values giving more (smaller) clusters.
Leiden
Leiden algorithm is applied iteratively, it converges to a partition in which all subsets of all communities are locally optimally assigned. Furthermore, by relying on a fast local move approach, the Leiden algorithm runs faster than the Louvain algorithm. The Leiden algorithm consists of three phases: (1) local moving of nodes, (2) refinement of the partition and (3) aggregation of the network based on the refined partition, using the non-refined partition to create an initial partition for the aggregate network.
Leiden Paper
SeuratObject <- FindClusters(
object = SeuratObject,
graph.name = "SNN",
resolution = 0.8,
algorithm = 4) # Setting `algorithm = 4` signifies Leiden
The number of clusters can be controlled using the resolution parameter with higher values giving more (smaller) clusters.
Hierarchical clustering
Hierarchical clustering (HC) is a method of cluster analysis which seeks to build a hierarchy of clusters. Strategies for hierarchical clustering generally fall into two types: Agglomerative or Divisive. […] In general, the merges and splits are determined in a greedy manner. The results of hierarchical clustering are usually presented in a dendrogram. […] The standard algorithm for hierarchical agglomerative clustering (HAC) has a time complexity of O(n3) and requires O(n2) memory, which makes it too slow for even medium data sets. In order to decide which clusters should be combined (for agglomerative), or where a cluster should be split (for divisive), a measure of dissimilarity between sets of observations is required. In most methods of hierarchical clustering, this is achieved by use of an appropriate metric (a measure of distance between pairs of observations), and a linkage criterion which specifies the dissimilarity of sets as a function of the pairwise distances of observations in the sets.
Wikipedia
HC for networks
The base R stats package already contains a function dist() that calculates distances between all pairs of samples. The distance methods available in dist() are: euclidean, maximum, manhattan, canberra, binary or minkowski. Additionally, we can also perform hierarchical clustering directly on a graph (KNN or SNN) which already contains information about cell-to-cell distances. However, since the distances in the graph are inverted (0s represent far and 1s represent close connections), we need to subtract from the maximum value on the graph (in the case of adjacency SNN, is 1), so that 0s represent close and 1s represent far distance.
After having calculated the distances between samples calculated, we can now proceed with the hierarchical clustering per-se. We will use the function hclust() for this purpose, in which we can simply run it with the distance objects created above. The methods available are: ward.D, ward.D2, single, complete, average, mcquitty, median or centroid. When clustering on a graph, use ward.D2.
# Running HC on a PCA
h <- hclust(
d = dist(SeuratObject@reductions[["pca"]]@cell.embeddings[, 1:30 ],
method = "euclidean"),
method = "ward.D2")
# Running HC on a graph
h <- hclust(
d = 1 - as.dist(SeuratObject@graphs$SNN),
method = "ward.D2",)
Once the cluster hierarchy is defined, the next step is to define which samples belong to a particular cluster.
plot(h, labels = FALSE)
After identifying the dendrogram for the cells (above), we can now literally cut the tree at a fixed threshold (with the cutree function) at different levels (a.k.a. resolutions) to define the clusters. The number of clusters can be controlled using the height (h) or directly via the k parameters.
# Cutting the tree based on a dendrogram height
SeuratObject$HC_res <- cutree(
tree = h,
k = 18)
# Cutting the tree based on a number of clusters
SeuratObject$HC_res <- cutree(
tree = h,
h = 3)
# To check how many cells are in each cluster
table(SeuratObject$HC_res)
The number of clusters can be controlled using the height (h) or directly via the k parameters.
K-mean
k-means clustering is a method of vector quantization, originally from signal processing, that aims to partition n observations into k clusters in which each observation belongs to the cluster with the nearest mean (cluster centers or cluster centroid), serving as a prototype of the cluster. […] The algorithm does not guarantee convergence to the global optimum. The result may depend on the initial clusters. As the algorithm is usually fast, it is common to run it multiple times with different starting conditions. […] k-means clustering tends to find clusters of comparable spatial extent (all with same size), while the expectation-maximization mechanism allows clusters to have different shapes.
K-means is a generic clustering algorithm that has been used in many application areas. In R, it can be applied via the kmeans function. Typically, it is applied to a reduced dimension representation of the expression data (most often PCA, because of the interpretability of the low-dimensional distances). We need to define the number of clusters in advance. Since the results depend on the initialization of the cluster centers, it is typically recommended to run K-means with multiple starting configurations (via the nstartargument).
set.seed(1)
SeuratObject$kmeans_12 <- kmeans(
x = SeuratObject@reductions[["pca"]]@cell.embeddings[, 1:50 ],
centers = 12,
iter.max = 50,
nstart = 10)$cluster
The number of clusters can be controlled using the centers parameter.
What clustering resolution should I use?
With all of these methods we can obtain any number of clusters by tweaking the settings. One of the hard problems in scRNA-seq analysis is to make reasonable decisions on how many clusters is reasonable. Unfortunately, there is no simple solution to this problem, it takes biological knowledge of the sample and some investigation to maker reasonable decisions on the number of clusters.
Differential gene expression may help in that analysis. If two clusters have the same DEGs and no clear genes that distinguish them, it may not be a good idea to split them into individual clusters. Note! Also examine the cluster you get with regards to the QC metrics (see above) to make sure that some clusters are not only formed due to low quality cells or doublets/multiples.
Comparing clusterings and metadata
A very useful task is to check how your clusters compare to other clustering conditions or metadata:
comparison_table <- table(list(
SeuratObject$meta.data[,"METADATA_FACTOR_1"],
SeuratObject$meta.data[,"METADATA_FACTOR_2"]
))
This will generate a table concatenating information of how many cells belong to the classes in comparison You can visualize it as a barplot, for example:
# Transform data to percentages:
comparison_table <- t(t(comparison_table) / colSums(comparison_table)) * 100
# Barplot
barplot(comparison_table,
col = scaleshue_pal()(nrow(comparison_table))[1:nrow(comparison_table)],
border = NA,
las = 2)
Differential expression
Once clusters have been defined, it is often informative to find the genes that define each cluster. The methods for DE prediction in scRNA-seq differs somewhat from bulk RNAseq methods. On one hand, we often have more samples (individual cells) compared to bulk RNA-seq, but on the other hand, the scRNA-seq data is noisy and suffer from drop-outs, which complicates things. You can read more about the different methods available in this paper.
Finding DEGs
Differentially expressed genes (DEGs) are often referred to as “marker genes”, however, you have to be aware that most DE tests are designed to detect genes that have higher expression in one group of cells compared to another. A DEG is not automatically a unique marker for a celltype.
The Seurat package has implemented many different tests for DE, some are designed for scRNA-seq and others are used also for bulk RNA-seq:
wilcox: Identifies differentially expressed genes between two groups of cells using a Wilcoxon Rank Sum testbimod: Likelihood-ratio test for single cell gene expression (McDavid et al., Bioinformatics, 2013)roc: Identifies ‘markers’ of gene expression using ROC analysis. For each gene, evaluates (using AUC) a classifier built on that gene alone, to classify between two groups of cells. An AUC value of 1 means that expression values for this gene alone can perfectly classify the two groupings (i.e. Each of the cells in cells.1 exhibit a higher level than each of the cells in cells.2). An AUC value of 0 also means there is perfect classification, but in the other direction. A value of 0.5 implies that the gene has no predictive power to classify the two groups. Returns a ‘predictive power’ (abs(AUC-0.5) * 2) ranked matrix of putative differentially expressed genes.t: Identify differentially expressed genes between two groups of cells using the Student’s t-test.negbinom: Identifies differentially expressed genes between two groups of cells using a negative binomial generalized linear model. Use only for UMI-based datasetspoisson: Identifies differentially expressed genes between two groups of cells using a poisson generalized linear model. Use only for UMI-based datasetsLR: Uses a logistic regression framework to determine differentially expressed genes. Constructs a logistic regression model predicting group membership based on each feature individually and compares this to a null model with a likelihood ratio test.MAST: Identifies differentially expressed genes between two groups of cells using a hurdle model tailored to scRNA-seq data. Utilizes the MAST package to run the DE testing.DESeq2: Identifies differentially expressed genes between two groups of cells based on a model using DESeq2 which uses a negative binomial distribution (Love et al, Genome Biology, 2014). This test does not support pre-filtering of genes based on average difference (or percent detection rate) between cell groups. However, genes may be pre-filtered based on their minimum detection rate (min.pct) across both cell groups. To use this method, please install DESeq2, using the instructions at [https://bioconductor.org/packages/release/bioc/html/DESeq2.html][]
To run DE prediction for all clusters in a Seurat object, each cluster vs. all other cells, use the FindAllMarkerfunction:
markers <- FindAllMarkers(
object = SeuratObject,
assay = "RNA",
logfc.threshold = 0.25,
test.use = "wilcox",
slot = "data",
min.pct = 0.1,
min.diff.pct = -Inf,
only.pos = FALSE,
max.cells.per.ident = Inf,
latent.vars = NULL,
min.cells.feature = 3,
min.cells.group = 3,
pseudocount.use = 1,
return.thresh = 0.01)
There are multiple cutoffs for including genes, filtering output etc that you can tweak. For instance only testing up-regulated genes may speed up the test:
- Only test up-regulated genes with
only.pos = TRUE - Minimum number of cells a gene is expressed in
min.cells.feature - Minimum number of cells of a cluster a gene is expressed in
min.cells.group - Only test genes with
min.pctexpression in a cluster - Only test genes with
min.pct.diffdifference in percent expression between two groups. - Return only genes with p.value <
return.thresh - Return only genes with logFoldchange >
logfc.threshold
Some other key features to think about:
- If you have multiple assays in your object, make sure to run DE on the correct assay. For instance, if you have integrated data, you still want to do DE on the “RNA” assay.
- If you have very uneven cluster sizes, it may bias the p-values of the genes so that clusters with many cells have more significant genes. It may be a good idea to set
max.cells.per.identto the size of your smallest cluster, and all clusters will be down-sampled to the same size. - Some of the tests allow you to include confounding factors in
latent.vars, those areLR, negbinom, poisson, or MAST.Comparing a cluster across experimental conditions
The second way of computing differential expression is to answer which genes are differentially expressed within a cluster. For example, we may have libraries coming from 2 different library preparation methods (batches) and we would like to know which genes are influenced the most in a particular cell type. The same concept applies if you have instead two or more biological groups (control vs treated, time#0 vs time#1 vs time#2, etc.).
For this end, we will first subset our data for the desired cell cluster, then change the cell identities to the variable of comparison (which now in our case is the “Batch”).
cell_selection <- SeuratObject[, SeuratObject$seurat_clusters == 4]
cell_selection <- SetIdent(cell_selection, value = "Batch")
# Compute differentiall expression
DGE_cell_selection <- FindAllMarkers(
object = cell_selection,
logfc.threshold = 0.2,
test.use = "wilcox",
min.pct = 0.1,
min.diff.pct = -Inf,
only.pos = FALSE,
max.cells.per.ident = Inf,
latent.vars = NULL,
min.cells.feature = 3,
min.cells.group = 3,
pseudocount.use = 1,
return.thresh = 0.01 )
We can also test any two set of cells using the function FindMarkers and specify the cell names for two groups as cells.1 and cells.2.
Plotting DEG results
Once we have ran a DE test, we may want to visualize the genes in different ways. But first, we need to get the top DEGs from each cluster. How to select top 5 genes per cluster:
top5 <- markers %>% group_by(cluster) %>% top_n(-5, p_val_adj)
These can be visualized in a heatmap:
DoHeatmap(object = SeuratObject,
features = as.character(unique(top5$gene)),
group.by = "seurat_clusters",
assay = "RNA")
Or with a dot-plot, where each cluster is represented with color by average expression, and size by proportion cells that have the gene expressed:
DotPlot(object = SeuratObject,
features = as.character(unique(top5$gene)),
group.by = "seurat_clusters",
assay = "RNA") +
coord_flip()
We can also plot a violin plot for each gene:
VlnPlot(object = SeuratObject,
features = as.character(unique(top5$gene)),
ncol = 5,
group.by = "seurat_clusters",
assay = "RNA")
The violin plot can also be split into batches, so if you have two batches with meta data column “Batch”, these can be plotted separately within each cluster for each gene. This may be very useful to check that the DEGs you have detected are not just driven by a single batch.
VlnPlot(object = SeuratObject,
features = as.character(unique(top5$gene)),
ncol = 5,
group.by = "seurat_clusters",
assay = "RNA",
split.by = "Batch")
Functional enrichment analysis
Having a defined list of enriched terms, you can now look for their combined function using enrichR:
# LOad additional packages
library(enrichR)
# Check available databases to perform enrichment (then choose one)
enrichR::listEnrichrDbs()
# Perform enrichment
enrich_results <- enrichr(
genes = top5$gene[top5$cluster == "CLUSTER_OF_INTEREST"],
databases = "DATABASE_OF_INTEREST" )[[1]]
Some databases of interest:
- GO_Biological_Process_2017b
- KEGG_2019_Human
- KEGG_2019_Mouse
- WikiPathways_2019_Human
- WikiPathways_2019_Mouse
You visualize your results using a simple barplot, for example:
mypar(1, 1, mar = c(3, 20, 2, 1))
barplot( height = -log10(en$P.value)[20:1],
names.arg = en$Term[20:1],
horiz = TRUE,
las = 2,
border = FALSE,
cex.names = .6 )
abline(v = c(-log10(0.05)), lty = 2)
abline(v = 0, lty = 1)
Trajectory inference
Below you can find a summary of data processing required for trajectory inference:
- A reduction where to perform the trajectory ( PCA > MDS > DM > UMAP > tSNE )
- The cell clustering information
Choosing the “best” method for trajectory inference is, like for most of the other scRNA-seq analyses non-trivial. For a comparison, please check this paper.
Trajectory inference with Slingshot
Slingshot is one of the most prototypical TI methods, as it contains many popular components that can also be found in other TI methods (Cannoodt, Saelens and Saeys 2016): dimensionality reduction, clustering and principal curves.
As many other TI methods, Slingshot is not restricted to a particular dimensionality reduction method. So which one should you use? There are three important points to take into account:
You should use enough dimensions to capture the whole complexity of the data. This is especially important for linear dimensionality reductions such as PCA and MDS, and if the trajectory topology is more complex than a bifurcation. Note that even when the trajectory is not clearly visible in two dimensions, the TI method may still see it in multiple dimensions. TI methods can see in a lot more dimensions compared to us silly earthlings!
- Some dimensionality reduction methods may enlarge (or blow up) the distance in high-density regions. Both t-SNE and UMAP (to a lesser extent) have this problem. The dataset that we have here doesn’t really have this problem, but it is quite common if you do an unbiased sampling of your biological populations.
- Some dimensionality reduction methods try to enforce a grouping of the cells, and remove continuities. Both t-SNE and UMAP (to a lesser extent) have this problem. These same methods may also put cells together that actually do not
- For these reasons, MDS and diffusion maps are often the preferred choice, although t-SNE and UMAP may work if you have a balanced and simple sample.
We want to find continuities in our data, why then do so many TI methods use methods that split up the data in distinct groups using clustering?dimred <- SeuratObject@reductions[['embedding_to_be_used']]@cell.embeddings
The answer is simple: clustering simplifies the problem by finding groups of cells that are at approximately the same location in the trajectory. These groups can then be connected to find the different paths that cells take, and where they branch off. Ideally, the clustering method for TI should therefore not group cells based on a density, such as DBSCAN A trajectory is by definition a group of cells that are all connected through high-density regions, but still differ in their expression.
We’ll use standard Seurat clustering here (louvain clustering), but alternative methods may be appropriate as well. Sometimes, it may be useful to “overcluster” to make sure that all sub-branches are captured. We’ll increase the resolution a bit, so that we find more granular clusters. As an exercise, you might tweak this resolution a bit and see how it affects downstream analyses.
clustering <- factor(SeuratObject@meta.data[, "clustering_to_be_used"])
Until up to this point, the steps above have been covered in the previous lectures. From now on, we will start using that clustering and data reduction techniques for trajectory inference. The whole process can be done using a single function named slingshot, which is simply a wrapper for the 2 main steps for trajectory inference. The first step of the process is to define the lineages and then fit a curve through the data that defines a trajectory. These steps are break down below for clarity.
Defining cell lineages with Slingshot
# Run default Slingshot lineage identification
library(slingshot)
set.seed(1)
lineages <- getLineages(data = dimred,
clusterLabels = clustering)
lineages
# Plot the lineages
par(mfrow = c(1,2))
plot(dimred[, 1:2],
col = clustering,
cex = .5,
pch = 16)
for(i in levels(clustering)) {
text(mean(dimred[clustering == i, 1]),
mean(dimred[clustering == i, 2]),
labels = i,
font = 2)
}
plot(dimred[, 1:2],
col = clustering,
cex = .5,
pch = 16)
lines(lineages,
lwd = 3,
col = 'black')
Here we see one central issue with trajectory analysis: where does the trajectory begin? Without any extra information, this is nearly an impossible task for a TI method. We need prior biological information to be able to define where the trajectory starts and where it should end.
# Run Slingshot starting at cluster 5
set.seed(1)
lineages <- getLineages(data = dimred,
clusterLabels = clustering,
start.clus = "5")
lineages
Defining Principal Curves
Once the clusters are connected, Slingshot allows you to transform them to a smooth trajectory using principal curves. This is an algorithm that iteratively changes an initial curve to better match the data points. It was developed for linear data. To apply it to single-cell data, slingshot adds two enhancements:
- It will run principal curves for each “lineage”, which is a set of clusters that go from a defined start cluster to some end cluster
- Lineages with a same set of clusters will be constrained so that their principal curves remain bundled around the overlapping clusters
Since the function getCurves() takes some time to run, we can speed up the convergence of the curve fitting process by reducing the amount of cells to use in each lineage. Ideally you could use all cells, but here we had set approx_points to 300 to speed up; feel free to adjust for your particular dataset.
curves <- getCurves(lineages,
approx_points = 300,
thresh = 0.01,
stretch = .8,
allow.breaks = FALSE,
shrink = .99)
curves
plot(dimred,
col = clustering,
asp = 1,
pch = 16)
lines(curves,
lwd = 3,
col = 'black')
Differential expression along trajectories
The main way to interpret a trajectory is to find genes that change along the trajectory. There are many ways to define differential expression along a trajectory, for example:
- Expression changes along a particular path (i.e. change with pseudotime)
- Expression differences between branches
- Expression changes at branch points
- Expression changes somewhere along the trajectory
tradeSeq is a recently proposed algorithm to find trajectory differentially expressed genes. It works by smoothing the gene expression along the trajectory by fitting a smoother using generalized additive models (GAMs), and testing whether certain coefficients are statistically different between points in the trajectory.
The fitting of GAMs can take quite a while, so for demonstration purposes we first do a very stringent filtering of the genes. In an ideal experiment, you would use all the genes, or at least those defined as being variable.
# Load additional packages
BiocParallel::register(BiocParallel::SerialParam())
library(tradeSeq)
# Remove some genes to speed up the computations for this tutorial
counts <- as.matrix(SeuratObject@assays$RNA@counts[SeuratObject@assays$RNA@var.features, ])
filt_counts <- counts[rowSums(counts > 5) > ncol(counts) / 100, ]
dim(filt_counts)
# Fitting a Gamma distribution
sce <- fitGAM(counts = as.matrix(filt_counts),
sds = curves )
plotGeneCount(curve = curves,
counts = filt_counts,
clusters = clustering,
models = sce)
Genes that change with pseudotime
pseudotime_association <- associationTest(sce)
pseudotime_association$fdr <- p.adjust(pseudotime_association$pvalue, method = "fdr")
pseudotime_association <- pseudotime_association[order(pseudotime_association$pvalue), ]
pseudotime_association$feature_id <- rownames(pseudotime_association)
# Function to plot differential expression
library(dplyr)
plot_differential_expression <- function(feature_id) {
cowplot::plot_grid(
plotGeneCount(curve = curves,
counts = filt_counts,
gene = feature_id[1],
clusters = clustering,
models = sce) +
ggplot2::theme(legend.position = "none"),
plotSmoothers(models = sce,
counts = as.matrix(counts),
gene = feature_id[1])
)}
# Get genes and plot
feature_id <- pseudotime_association %>%
filter(pvalue < 0.05) %>%
top_n(1, -waldStat) %>%
pull(feature_id)
plot_differential_expression(feature_id)
Genes that change between two pseudotime points
We can define custom pseudotime values of interest if we’re interested in genes that change between particular point in pseudotime (for example, the progenitor cell markers). By default, we can look at differences between start and end:
pseudotime_start_end_association <- startVsEndTest(sce, pseudotimeValues = c(0, 1))
pseudotime_start_end_association$feature_id <- rownames(pseudotime_start_end_association)
feature_id <- pseudotime_start_end_association %>%
filter(pvalue < 0.05) %>%
top_n(1, waldStat) %>%
pull(feature_id)
plot_differential_expression(feature_id)
Genes that are different between lineages
More interesting are genes that are different between two branches. We may have seen some of these genes already pop up in previous analyses of pseudotime. There are several ways to define “different between branches”, and each have their own functions:
- Different at the end points between lineages, using
diffEndTest - Different at the branching point between lineages, using
earlyDETest - Different somewhere in pseudotime the branching point, using
patternTest
Note that the last function requires that the pseudotimes between two lineages are aligned.
different_end_association <- diffEndTest(sce)
different_end_association$feature_id <- rownames(different_end_association)
feature_id <- different_end_association %>%
filter(pvalue < 0.05) %>%
arrange(desc(waldStat)) %>%
dplyr::slice(1) %>%
pull(feature_id)
plot_differential_expression(feature_id)
branch_point_association <- earlyDETest(sce)
branch_point_association$feature_id <- rownames(branch_point_association)
feature_id <- branch_point_association %>%
filter(pvalue < 0.05) %>%
arrange(desc(waldStat)) %>%
dplyr::slice(1) %>%
pull(feature_id)
plot_differential_expression(feature_id)
Check out this vignette for a more in-depth overview of tradeSeq
References
Saelens, Wouter, Robrecht Cannoodt, Helena Todorov, and Yvan Saeys. 2019. “A Comparison of Single-Cell Trajectory Inference Methods.” Nature Biotechnology 37 (5): 547–54. doi.
Additional material
The field of scRNA-seq is both large and continuously evolving - there is thus a slew of available methods, tools and software to do both varied and similar things with. Here you we list some additional materials that you might find useful to explore to get an even deeper understanding.
Online courses
- Exercises for the NBIS scRNA-seq course
- The GitHub repository for the NBIS scRNA-seq course
- Single cell RNA-seq course at from Hemberg lab
- Single cell RNA-seq course in Python
Single cell RNA-seq course at Broad
Presentations and lectures
- Quaility controls
- Normalisation
- Dimensionality reduction
- Batch correction and data integration
- Clustering techniques
- Differential expression analysis
- Trajectory inference analyses
All of the above in a YouTube playlist
Tools and pipelines
- Bitbucket repository for QC report scripts
- Bitbucket repository for an NBIS scRNA-seq pipeline
A catalogue of many scRNA-seq tools
Websites
- Conquer datasets - many different datasets based on a salmon pipeline
- The Human Cell Atlas project
The EBI Single-cell expression atlas
Additional papers
- A systematic evaluation of single cell RNA-seq analysis pipelines
Source:https://nbisweden.github.io/single-cell-pbl/glossary_of_terms_single_cell.html#

若有收获,就点个赞吧
0 人点赞
