
在本教程中,我们将学习Seurat包中进行差异表达分析寻找marker基因的常用方法。
加载所需的R包和数据集
library(Seurat)# 这里我们使用之前分析用过的PBMC数据pbmc <- readRDS(file = "../data/pbmc3k_final.rds")
使用默认的方法进行差异表达分析
使用FindMarkers函数进行差异表达分析。默认情况下,FindMarkers函数使用非参数的Wilcoxon秩和检验进行差异表达分析。如果要对两组特定的细胞类群执行差异分析,可以设置ident.1和ident.2参数指定两个特定的细胞类群。
# list options for groups to perform differential expression on# 查看已分群好的细胞类群levels(pbmc)[1] "Naive CD4 T" "Memory CD4 T" "CD14+ Mono" "B" "CD8 T"[6] "FCGR3A+ Mono" "NK" "DC" "Platelet"# Find differentially expressed features between CD14+ and FCGR3A+ Monocytes# 使用默认方法对CD14+ and FCGR3A+ Monocytes两组细胞类群进行差异表达分析monocyte.de.markers <- FindMarkers(pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono")# 查看筛选出的一些marker基因head(monocyte.de.markers)
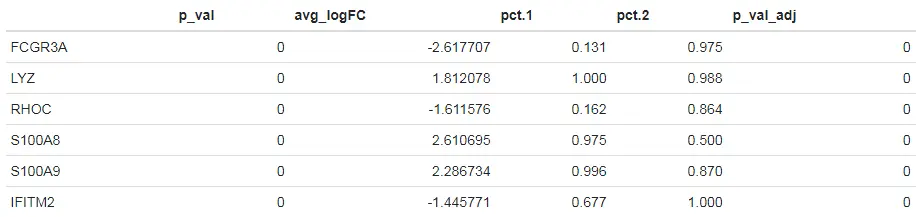
上述结果表格中主要包含以下列:
- p_val:假设检验后得到的原始P值
- avg_logFC:两组之间平均表达差异倍数的对数值。正值表示该基因在第一组中的表达更高。
- pct.1:第一组中检测到表达该基因的细胞所占的百分比
- pct.2:第二组中检测到表达该基因的细胞所占的百分比
- p_val_adj:bonferroni多重检验校正后得到的校正后的P值。
如果省略了ident.2参数或将其设置为NULL,FindMarkers函数将对指定的ident.1组与其他所有组之间进行差异表达分析。
# Find differentially expressed features between CD14+ Monocytes and all other cells, only search for positive markersmonocyte.de.markers <- FindMarkers(pbmc, ident.1 = "CD14+ Mono", ident.2 = NULL, only.pos = TRUE)# view resultshead(monocyte.de.markers)
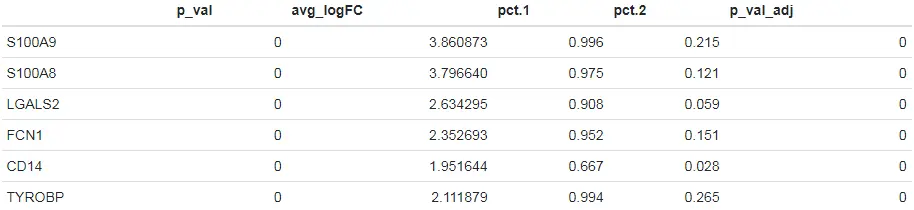
在差异表法分析时预过滤一些基因或细胞可提高DE测试的速度
为了提高差异表达分析的速度,特别是对于那些较大的数据集,Seurat允许在进行差异表达分析时对基因或细胞进行预过滤。例如,在一组细胞中很少检测到的基因,或具有相似的平均表达水平的基因不太可能被差异表达。我们可以通过以下参数min.pct,logfc.threshold,min.diff.pct,和max.cells.per.ident进行基因或细胞的预过滤。
# Pre-filter features that are detected at <50% frequency in either CD14+ Monocytes or FCGR3A+ Monocytes# 设置min.pct = 0.5参数过滤掉那些在50%以下细胞中检测到的基因head(FindMarkers(pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono", min.pct = 0.5))
# Pre-filter features that have less than a two-fold change between the average expression of CD14+ Monocytes vs FCGR3A+ Monocytes# 设置logfc.threshold = log(2)参数过滤掉那些在两个不同组之间平均表达的差异倍数低于2的基因head(FindMarkers(pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono", logfc.threshold = log(2)))
# Pre-filter features whose detection percentages across the two groups are similar (within 0.25)# 设置min.diff.pct = 0.25参数过滤掉那些在两个不同组之间能检测到的细胞比例低于0.25的基因head(FindMarkers(pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono", min.diff.pct = 0.25))
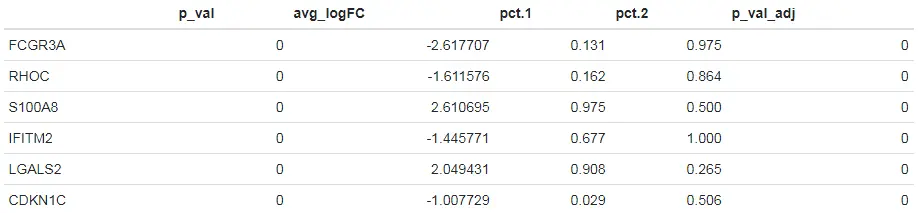
# Increasing min.pct, logfc.threshold, and min.diff.pct, will increase the speed of DE testing, but could also miss features that are prefiltered# Subsample each group to a maximum of 200 cells. Can be very useful for large clusters, or computationally-intensive DE testshead(FindMarkers(pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono", max.cells.per.ident = 200))
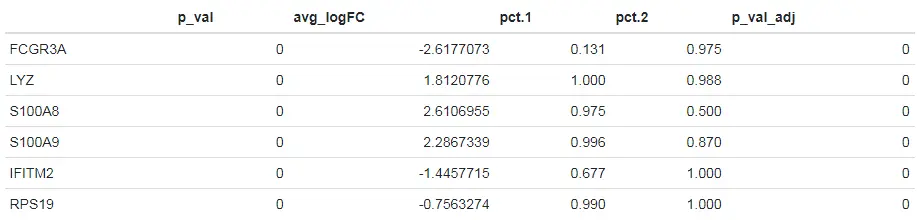
使用其他的检验方法进行差异表达分析
目前,Seurat中主要支持以下差异表达检验的方法:
- “wilcox” : Wilcoxon rank sum test (default)
- “bimod” : Likelihood-ratio test for single cell feature expression, (McDavid et al., Bioinformatics, 2013)
- “roc” : Standard AUC classifier
- “t” : Student’s t-test
- “poisson” : Likelihood ratio test assuming an underlying poisson distribution. Use only for UMI-based datasets
- “negbinom” : Likelihood ratio test assuming an underlying negative binomial distribution. Use only for UMI-based datasets
- “LR” : Uses a logistic regression framework to determine differentially expressed genes. Constructs a logistic regression model predicting group membership based on each feature individually and compares this to a null model with a likelihood ratio test.
- “MAST” : GLM-framework that treates cellular detection rate as a covariate (Finak et al, Genome Biology, 2015)
- “DESeq2” : DE based on a model using the negative binomial distribution (Love et al, Genome Biology, 2014)
对于MAST和DESeq2方法,我们需要单独安装这些软件包,以便将它们用于Seurat的差异表达分析中。安装后,可以通过test.use参数指定要使用的DE检验方法。
# Test for DE features using the MAST package# 设置test.use = "MAST"参数指定使用MAST方法head(FindMarkers(pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono", test.use = "MAST"))
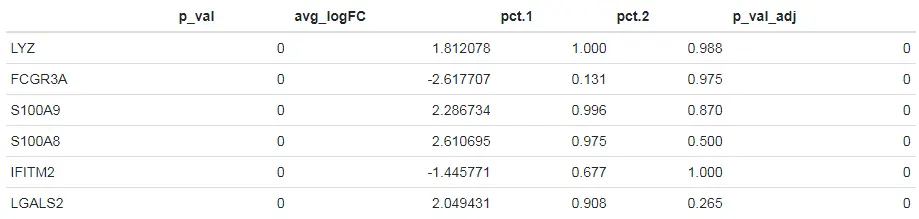
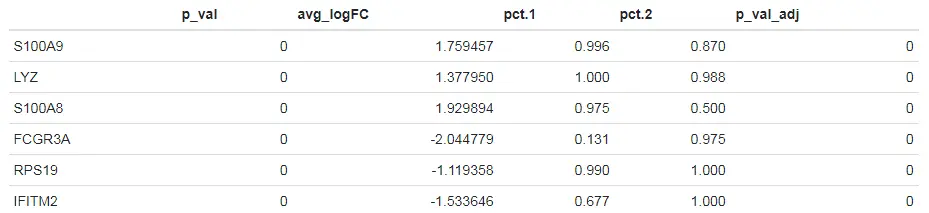
# Test for DE features using the DESeq2 package. Throws an error if DESeq2 has not already been installed Note that the DESeq2 workflows can be computationally intensive for large datasets, but are incompatible with some feature pre-filtering options We therefore suggest initially limiting the number of cells used for testing# 设置test.use = "DESeq2"参数指定使用DESeq2方法head(FindMarkers(pbmc, ident.1 = "CD14+ Mono", ident.2 = "FCGR3A+ Mono", test.use = "DESeq2", max.cells.per.ident = 50))

若有收获,就点个赞吧
0 人点赞
