

往期精选
使用Signac包进行单细胞ATAC-seq数据分析(一):Analyzing PBMC scATAC-seq
使用Signac包进行单细胞ATAC-seq数据分析(二):Motif analysis with Signac
使用Signac包进行单细胞ATAC-seq数据分析(三):scATAC-seq data integration
使用Signac包进行单细胞ATAC-seq数据分析(四):Merging objects
Cicero是一个用于分析
单细胞染色质可及性实验的R工具包。Cicero的主要功能是使用单细胞染色质可及性数据,通过分析共同可及性来预测基因组中的顺式调控相互作用(如增强子和启动子之间的相互作用)。此外,Cicero包还扩展了Monocle的功能,能够利用染色质可及性数据对单细胞进行聚类,排序和差异可及性分析。
Cicero包的简介
Cicero包的主要功能是使用单细胞染色质可及性数据来预测基因组中更可能位于细胞核附近的区域。这可用于鉴定潜在的增强子-启动子互作对,并了解全基因组区域内的顺式相互作用的整体结构。
由于单细胞数据的稀疏性,细胞必须根据相似度进行聚合,以对数据中的各种技术因素进行可靠的校正。最终,Cicero根据用户给定的距离,计算在指定距离内每个可及性peaks对之间的开放性,并给出”Cicero co-accessibility”得分,其分数介于-1和1之间,得分越高,表示更高的共可及性(co-accessibility)。
此外,Cicero包还提供了扩展工具包,可使用Monocle提供的框架来分析单细胞ATAC-seq数据。
Cicero包提供了两种主要的分析功能:
- 构建和分析顺式调控网络。 Cicero通过分析共可及性(co-accessibility),来识别鉴定潜在的顺式调控相互作用,并使用各种技术对其进行可视化和分析。
- 常规单细胞染色质可及性分析。 Cicero还扩展了Monocle包,以使用单细胞染色质可及性数据来进行差异可及性(differential accessibility)分析,细胞聚类与可视化,和发育轨迹重建。
Cicero包的安装
通过Bioconductor进行安装
if (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager")# 安装依赖包BiocManager::install(c("Gviz", "GenomicRanges", "rtracklayer"))BiocManager::install("cicero")# 加载cicero包library(cicero)
通过Github进行安装
if (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager")# 安装依赖包BiocManager::install(c("Gviz", "GenomicRanges", "rtracklayer"))install.packages("devtools")devtools::install_github("cole-trapnell-lab/cicero-release")# 加载cicero包library(cicero)
数据集的加载
Cicero将数据存储在CellDataSet(CDS)类的对象中,该类继承自Bioconductor的ExpressionSet类。我们可以使用以下三个函数来操作该对象:
- fData: 获取feature的元信息
- pData: 获取cell/sample的元信息
- exprs: 获取cell-by-peak的count矩阵
为了修改CDS对象以保留染色质可及性数据而不是表达数据,Cicero使用peaks作为feature数据而不是基因或转录本。具体来说,许多Cicero函数需要形式为chr1_10390134_10391134的peaks值信息,如下所示: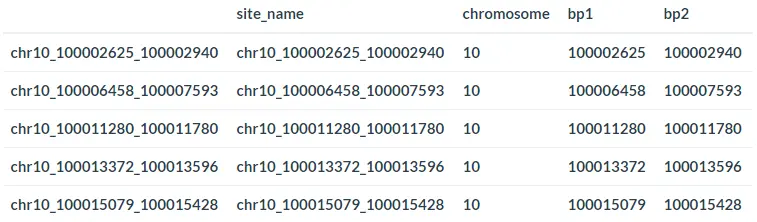
Loading data from a simple sparse matrix format
Cicero可以读取以简单稀疏矩阵格式存储的数据,这里以Cicero包自带的一个小数据集cicero_data为例。
# 加载示例数据集data(cicero_data)# 查看示例数据head(cicero_data)Peak Cell Count140 chr18_30209631_30210783 AGCGATAGGCGCTATGGTGGAATTCAGTCAGGACGT 4150 chr18_45820294_45821666 AGCGATAGGTAGCAGCTATGGTAATCCTAGGCGAAG 2185 chr18_32820116_32820994 TAATGCGCCGCTTATCGTTGGCAGCTCGGTACTGAC 2266 chr18_41888433_41890138 AGCGATAGGCGCTATGGTGGAATTCAGTCAGGACGT 2273 chr18_33038287_33039444 AGCGATAGGGTTATCGAACTCCATCGAGGTACTGAC 2285 chr18_25533921_25534483 ATTACTCGAACGCGCAGAGGCGGAGGTCGTACTGAC 1
为了方便起见,Cicero提供了一个名为make_atac_cds的函数。该函数以稀疏矩阵格式将data.frame或文件的路径作为输入。具体来说,此文件应该是由三列组成的制表符分隔的文本文件。 第一列是peak的坐标,格式为“ chr10_100013372_100013596”,第二列是细胞的名称,第三列是整数,表示细胞在该peak重叠的读取次数,且此文件不应包含标题行,如下所示: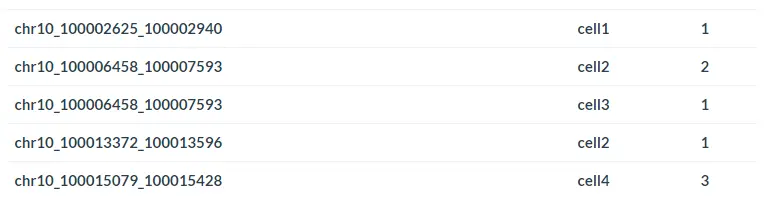
# 使用make_atac_cds函数将稀疏矩阵转换为CDS对象input_cds <- make_atac_cds(cicero_data, binarize = TRUE)# 查看CDS对象input_cdsCellDataSet (storageMode: environment)assayData: 6146 features, 200 sampleselement names: exprsprotocolData: nonephenoDatasampleNames: AGCGATAGAACGAATTCGGCGCAATGACCCTATCCTAGCGATAGAAGTACGCGATCCGCGGACTGTACTGAC ...TCTCGCGCTCTTGAGGTTTTATGACCAAATAGAGGC (200 total)varLabels: cells Size_Factor num_genes_expressedvarMetadata: labelDescriptionfeatureDatafeatureNames: chr18_10025_10225 chr18_10603_11103 ...chr18_78015362_78016311 (6146 total)fvarLabels: site_name chr ... num_cells_expressed (5 total)fvarMetadata: labelDescriptionexperimentData: use 'experimentData(object)'Annotation:# 查看feature的metadatahead(fData(input_cds))site_name chr bp1 bp2chr18_10025_10225 chr18_10025_10225 18 10025 10225chr18_10603_11103 chr18_10603_11103 18 10603 11103chr18_11604_13986 chr18_11604_13986 18 11604 13986chr18_49557_50057 chr18_49557_50057 18 49557 50057chr18_50240_50740 chr18_50240_50740 18 50240 50740chr18_104385_104585 chr18_104385_104585 18 104385 104585num_cells_expressedchr18_10025_10225 5chr18_10603_11103 1chr18_11604_13986 9chr18_49557_50057 2chr18_50240_50740 2chr18_104385_104585 1# 查看cell的metadatahead(pData(input_cds))cellsAGCGATAGAACGAATTCGGCGCAATGACCCTATCCT AGCGATAGAACGAATTCGGCGCAATGACCCTATCCTAGCGATAGAAGTACGCGATCCGCGGACTGTACTGAC AGCGATAGAAGTACGCGATCCGCGGACTGTACTGACAGCGATAGAATACGATAAGGCCGTCAACTAATCTTA AGCGATAGAATACGATAAGGCCGTCAACTAATCTTAAGCGATAGATTATGCAAGCCAGTACTTGCCTATCCT AGCGATAGATTATGCAAGCCAGTACTTGCCTATCCTAGCGATAGCAGACTAAGGGGAATTCAGTGGCTCTGA AGCGATAGCAGACTAAGGGGAATTCAGTGGCTCTGAAGCGATAGCCGTATGATTAGATCTTGGTCAGGACGT AGCGATAGCCGTATGATTAGATCTTGGTCAGGACGTSize_Factor num_genes_expressedAGCGATAGAACGAATTCGGCGCAATGACCCTATCCT NA 290AGCGATAGAAGTACGCGATCCGCGGACTGTACTGAC NA 490AGCGATAGAATACGATAAGGCCGTCAACTAATCTTA NA 253AGCGATAGATTATGCAAGCCAGTACTTGCCTATCCT NA 181AGCGATAGCAGACTAAGGGGAATTCAGTGGCTCTGA NA 85AGCGATAGCCGTATGATTAGATCTTGGTCAGGACGT NA 251# 查看cell-by-peak的count矩阵head(exprs(input_cds))6 x 200 sparse Matrix of class "dgCMatrix"[[ suppressing 32 column names 'AGCGATAGAACGAATTCGGCGCAATGACCCTATCCT', 'AGCGATAGAAGTACGCGATCCGCGGACTGTACTGAC', 'AGCGATAGAATACGATAAGGCCGTCAACTAATCTTA' ... ]]chr18_10025_10225 . . . . . . . . . . . . . . . . . . . . . . . . . . . .chr18_10603_11103 . . . . . . . . . . . . . . . . . . . . . . . . . . . .chr18_11604_13986 . 1 . . . . . . . . . . . . . . . . . . . . . . . . . .chr18_49557_50057 . . 1 . . . . . . . . . . . . . . . . . . . . . . . . .chr18_50240_50740 . . . . . . . . . . . . . . . . . . . . . . . . . . . .chr18_104385_104585 . . . . . . . . . . . . . . . . . . . . . . . . . . . .chr18_10025_10225 . . . . ......chr18_10603_11103 . . . . ......chr18_11604_13986 . . . . ......chr18_49557_50057 . . . . ......chr18_50240_50740 . . . . ......chr18_104385_104585 . 1 . . ...........suppressing 168 columns in show(); maybe adjust 'options(max.print= *, width = *)'..............................
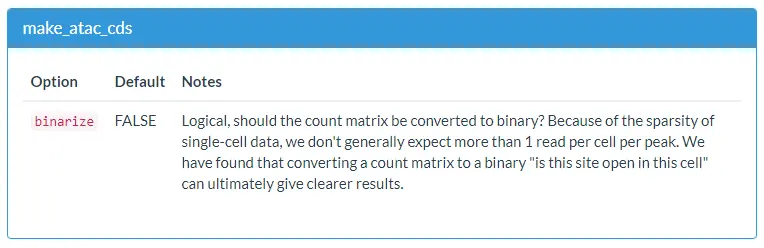
Loading 10X scATAC-seq data
如果我们的scATAC-seq数据来自10x Genomics平台,使用Cell Ranger ATAC软件处理将数据输出到一个名为filtered_peak_bc_matrix的文件夹中,可以通过以下方式将数据转换为CDS对象。
加载cell-by-peak的count矩阵# read in matrix data using the Matrix packageindata <- Matrix::readMM("filtered_peak_bc_matrix/matrix.mtx")# binarize the matrixindata@x[indata@x > 0] <- 1# 加载cell的metadata# format cell infocellinfo <- read.table("filtered_peak_bc_matrix/barcodes.tsv")row.names(cellinfo) <- cellinfo$V1names(cellinfo) <- "cells"# 加载peak的metadata# format peak infopeakinfo <- read.table("filtered_peak_bc_matrix/peaks.bed")names(peakinfo) <- c("chr", "bp1", "bp2")peakinfo$site_name <- paste(peakinfo$chr, peakinfo$bp1, peakinfo$bp2, sep="_")row.names(peakinfo) <- peakinfo$site_namerow.names(indata) <- row.names(peakinfo)colnames(indata) <- row.names(cellinfo)# 使用newCellDataSet函数构建CDS对象# make CDSfd <- methods::new("AnnotatedDataFrame", data = peakinfo)pd <- methods::new("AnnotatedDataFrame", data = cellinfo)input_cds <- suppressWarnings(newCellDataSet(indata,phenoData = pd,featureData = fd,expressionFamily=VGAM::binomialff(),lowerDetectionLimit=0))input_cds@expressionFamily@vfamily <- "binomialff"input_cds <- monocle::detectGenes(input_cds)# 数据初步过滤#Ensure there are no peaks included with zero readsinput_cds <- input_cds[Matrix::rowSums(exprs(input_cds)) != 0,]
参考来源:https://cole-trapnell-lab.github.io/cicero-release/docs/

若有收获,就点个赞吧
0 人点赞
