Generating FASTQs with cellranger-atac mkfastq
Overview
cellranger-atac软件提供了cellranger-atac mkfastq子程序,可以将Illumina测序仪产生的原始base call files(BCLs)下机数据转换为FASTQ格式的测序文件。该子程序是bcl2fastq软件的封装,提供了以下功能:
- Translates
10x sample index set namesinto the corresponding list offour sample index oligonucleotides. For example, well A1 can be specified in the samplesheet as SI-NA-A1, andcellranger-atac mkfastqwill recognize thefour oligosAAACGGCG, CCTACCAT, GGCGTTTC, and TTGTAAGA andmerge the resulting FASTQ files. - Supports a
simplified CSV samplesheetformat to handle 10x use cases. - Generates sequencing and 10x-specific
quality control metrics, including barcode quality, accuracy, and diversity. - Supports most
bcl2fastq arguments, such as--use-bases-mask.
Example Workflows
在本示例中,我们构建了两个10x测序文库(each processed through a separate Chromium chip channel),然后将它们混合在一起在同一个flowcell上进行测序。使用cellranger-atac mkfastq子程序拆库分离后,对每个测序文库的数据进行单独处理。
在本示例中,我们构建了一个10x测序文库,然后将它们放到两个不同的flowcell上进行测序。使用cellranger-atac mkfastq子程序将数据转换为FASTQ格式后,把它们合并到一起进行处理。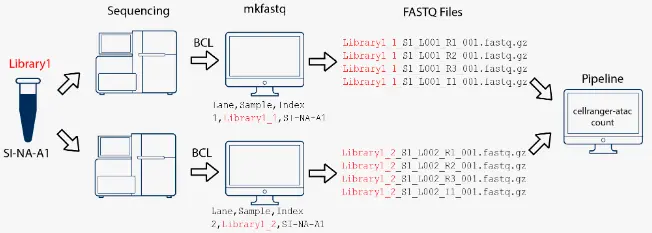
Arguments and Options
cellranger-atac mkfastq子程序是bcl2fastq软件的封装,主要提供了以下常用参数:

Example Data
cellranger-atac mkfastq子程序可以识别两种格式的文件进行处理:一种是常见的逗号分隔的3列格式的CSV文件;另一种是Illumina® Experiment Manager (IEM)格式的文件。
To follow along, please do the following:
Download the tiny-bcltar file.Untar the tiny-bcltar file in a convenient location. This will create a new tiny-bcl subdirectory.- Download the
simple CSV layout file: cellranger-atac-tiny-bcl-simple-1.0.0.csv. - Download the
Illumina® Experiment Manager sample sheet: cellranger-atac-tiny-bcl-samplesheet-1.0.0.csv.
Running mkfastq with a simple CSV samplesheet
我们推荐使用简单的CSV格式文件进行处理,该文件包含三列,分别是(Lane, Sample, Index)
Lane,Sample,Index1,test_sample,SI-NA-C1
Here are the options for each column:
To run cellranger-atac mkfastq with a simple layout CSV, use the --csv argument. Here’s how to run mkfastq on the tiny-bcl sequencing run with the simple layout:
cellranger-atac mkfastq --id=tiny-bcl \--run=/path/to/tiny_bcl \--csv=cellranger-atac-tiny-bcl-simple-1.0.0.csvcellranger-atac mkfastqCopyright (c) 2018 10x Genomics, Inc. All rights reserved.-------------------------------------------------------------------------------Martian Runtime - 1.2.0-3.2.4Running preflight checks (please wait)...2018-08-09 16:33:54 [runtime] (ready) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET2018-08-09 16:33:57 [runtime] (split_complete) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET2018-08-09 16:33:57 [runtime] (run:local) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET.fork0.chnk0.main2018-08-09 16:34:00 [runtime] (chunks_complete) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET...
Running mkfastq with an Illumina® Experiment Manager sample sheet
cellranger-atac mkfastq子程序还可以使用Illumina® Experiment Manager (IEM)格式的样本表作为输入。
Let’s briefly look at cellranger-atac-tiny-bcl-samplesheet-1.0.0.csv before running the pipeline. You will see a number of fields specific to running on Illumina® platforms, and then a [Data] section.
That section is where to put your sample, lane and index information. Here’s an example:
[Data]Lane,Sample_ID,index,Sample_Project1,Sample1,SI-NA-C1,tiny-bcl
Next, run the cellranger-atac mkfastq pipeline, using the --samplesheet argument:
cellranger-atac mkfastq --id=tiny-bcl \--run=/path/to/tiny_bcl \--samplesheet=cellranger-atac-tiny-bcl-samplesheet-1.0.0.csvcellranger-atac mkfastqCopyright (c) 2018 10x Genomics, Inc. All rights reserved.-------------------------------------------------------------------------------Martian Runtime - 3.2.4Running preflight checks (please wait)...2018-08-09 16:25:49 [runtime] (ready) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET2018-08-09 16:25:52 [runtime] (split_complete) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET2018-08-09 16:25:52 [runtime] (run:local) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET.fork0.chnk0.main2018-08-09 16:25:58 [runtime] (chunks_complete) ID.tiny-bcl.MAKE_FASTQS_CS.MAKE_FASTQS.PREPARE_SAMPLESHEET...
Checking FASTQ output
运行完cellranger-atac mkfastq程序后,会生成一个新的文件夹,存放着转换好的FASTQ文件,该文件夹名是--id参数指定的名称(if not specified, defaults to the name of the flowcell):
$ ls -ldrwxr-xr-x 4 jdoe jdoe 4096 Sep 13 12:05 tiny-bcl
The key output files can be found in outs/fastq_path, and is organized in the same manner as a conventional bcl2fastq run:
$ ls -l tiny-bcl/outs/fastq_path/drwxr-xr-x 3 jdoe jdoe 3 Aug 9 12:26 Reportsdrwxr-xr-x 2 jdoe jdoe 8 Aug 9 12:26 Statsdrwxr-xr-x 3 jdoe jdoe 3 Aug 9 12:26 tiny-bcl-rw-r--r-- 1 jdoe jdoe 20615106 Aug 9 12:26 Undetermined_S0_L001_I1_001.fastq.gz-rw-r--r-- 1 jdoe jdoe 151499694 Aug 9 12:26 Undetermined_S0_L001_R1_001.fastq.gz-rw-r--r-- 1 jdoe jdoe 52692701 Aug 9 12:26 Undetermined_S0_L001_R2_001.fastq.gz-rw-r--r-- 1 jdoe jdoe 151499694 Aug 9 12:26 Undetermined_S0_L001_R3_001.fastq.gz$ tree tiny-bcl/outs/fastq_path/tiny_bcl/tiny-bcl/outs/fastq_path/tiny_bcl/Sample1Sample1_S1_L001_I1_001.fastq.gzSample1_S1_L001_R1_001.fastq.gzSample1_S1_L001_R2_001.fastq.gzSample1_S1_L001_R3_001.fastq.gz
If you want to remove the Undetermined FASTQs from the output to save space, you can run mkfastq with the --delete-undetermined flag.
Assessing Quality Control Metrics
When the --qc flag is specified, the cellranger-atac mkfastq pipeline writes both sequencing and 10x-specific quality control metrics into a JSON file. The metrics are in the outs/qc_summary.json file.
The use of --qc flag is not supported on NovaSeq flow cells.
The qc_summary.json file contains a number of useful metrics. The sample_qc key is a good place to start exploring your data.
"sample_qc": {"Sample1": {"5": {"barcode_exact_match_ratio": 0.9336158258904611,"barcode_q30_base_ratio": 0.9611993091728814,"bc_on_whitelist": 0.9447542078230667,"mean_barcode_qscore": 37.770630795934,"number_reads": 2748155,"read1_q30_base_ratio": 0.8947676653366835,"read2_q30_base_ratio": 0.7771883245304577},"all": {"barcode_exact_match_ratio": 0.9336158258904611,"barcode_q30_base_ratio": 0.9611993091728814,"bc_on_whitelist": 0.9447542078230667,"mean_barcode_qscore": 37.770630795934,"number_reads": 2748155,"read1_q30_base_ratio": 0.8947676653366835,"read2_q30_base_ratio": 0.7771883245304577}}}
The sample_qc metric is a series of key value pairs for each sample in the sample sheet, and one metrics structure per lane per sample, plus an ‘all’ structure in case a sample spans multiple lanes.
The metrics are as follows: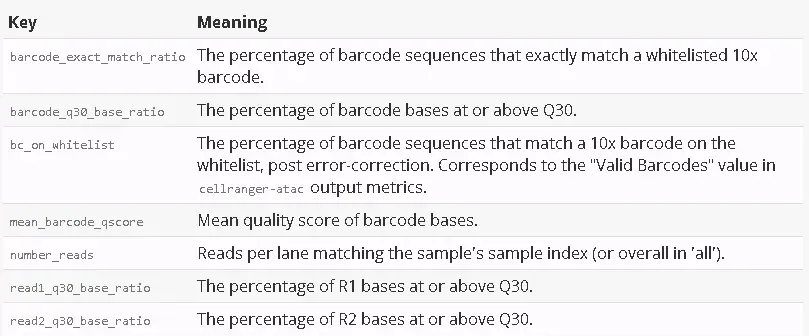
Single-Library Analysis with cellranger-atac count
Cell Ranger ATAC's pipelines analyze sequencing data produced from Chromium Single Cell ATAC libraries. This involves the following steps:
- Run
cellranger-atac mkfastqon the Illumina® BCL output folder to generate FASTQ files. - Run
cellranger-atac count on each library that was demultiplexed by cellranger-atac mkfastq.
Run cellranger-atac count
To generate single-cell accessibility counts for a single library, run cellranger-atac count with the following arguments. For a complete list of command-line arguments, run cellranger-atac count --help.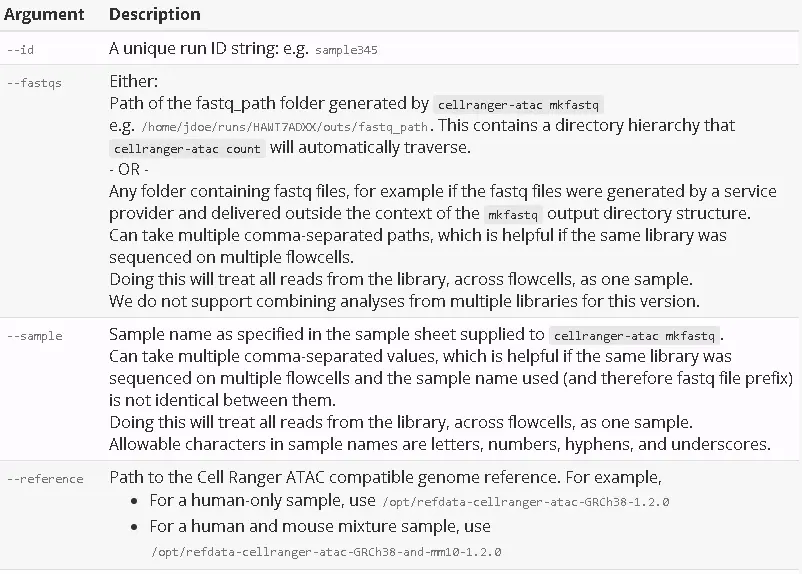

After determining these input arguments, run cellranger-atac:
$ cd /home/jdoe/runs$ cellranger-atac count --id=sample345 \--reference=/opt/refdata-cellranger-atac-GRCh38-1.2.0 \--fastqs=/home/jdoe/runs/HAWT7ADXX/outs/fastq_path \--sample=mysample \--localcores=8 \--localmem=64Martian Runtime - 3.2.4Running preflight checks (please wait)...2018-09-17 21:33:47 [runtime] (ready) ID.sample345.SC_ATAC_COUNTER_CS.SC_ATAC_COUNTER._BASIC_SC_ATAC_COUNTER._ALIGNER.SETUP_CHUNKS2018-09-17 21:33:47 [runtime] (run:local) ID.sample345.SC_ATAC_COUNTER_CS.SC_ATAC_COUNTER._BASIC_SC_ATAC_COUNTER._ALIGNER.SETUP_CHUNKS.fork0.chnk0.main2018-09-17 21:33:56 [runtime] (chunks_complete) ID.sample345.SC_ATAC_COUNTER_CS.SC_ATAC_COUNTER._BASIC_SC_ATAC_COUNTER._ALIGNER.SETUP_CHUNKS...
默认情况下,cellranger-atac count子程序会调用系统中可用的所有CPU进行计算,我们可以通过设置--localcores参数指定要用的CPU个数,如设置—localcores=16参数指定使用16个CPU进行计算。同样的,我们还可以通过设置--localmem参数指定要使用的运行内存。
Output Files
A successful cellranger-atac count run should conclude with a message similar to this:
2018-09-17 22:26:56 [runtime] (join_complete) ID.sample345.SC_ATAC_COUNTER_CS.SC_ATAC_COUNTER.CLOUPE_PREPROCESSOutputs:- Per-barcode fragment counts & metrics: /opt/sample345/outs/singlecell.csv- Position sorted BAM file: /opt/sample345/outs/possorted_bam.bam- Position sorted BAM index: /opt/sample345/outs/possorted_bam.bam.bai- Summary of all data metrics: /opt/sample345/outs/summary.json- HTML file summarizing data & analysis: /opt/sample345/outs/web_summary.html- Bed file of all called peak locations: /opt/sample345/outs/peaks.bed- Raw peak barcode matrix in hdf5 format: /opt/sample345/outs/raw_peak_bc_matrix.h5- Raw peak barcode matrix in mex format: /opt/sample345/outs/raw_peak_bc_matrix- Directory of analysis files: /opt/sample345/outs/analysis- Filtered peak barcode matrix in hdf5 format: /opt/sample345/outs/filtered_peak_bc_matrix.h5- Filtered peak barcode matrix: /opt/sample345/outs/filtered_peak_bc_matrix- Barcoded and aligned fragment file: /opt/sample345/outs/fragments.tsv.gz- Fragment file index: /opt/sample345/outs/fragments.tsv.gz.tbi- Filtered tf barcode matrix in hdf5 format: /opt/sample345/outs/filtered_tf_bc_matrix.h5- Filtered tf barcode matrix in mex format: /opt/sample345/outs/filtered_tf_bc_matrix- Loupe Cell Browser input file: /opt/sample345/outs/cloupe.cloupe- csv summarizing important metrics and values: /opt/sample345/outs/summary.csvPipestance completed successfully!
运行完cellranger-atac coun子程序后,会生成一个sample ID名的新文件夹,里面存放着分析好的结果文件。我们可以使用浏览器查看summary HTML文件,还可以使用Loupe Cell Browser查看生成的.cloupe文件进行可视化的探索。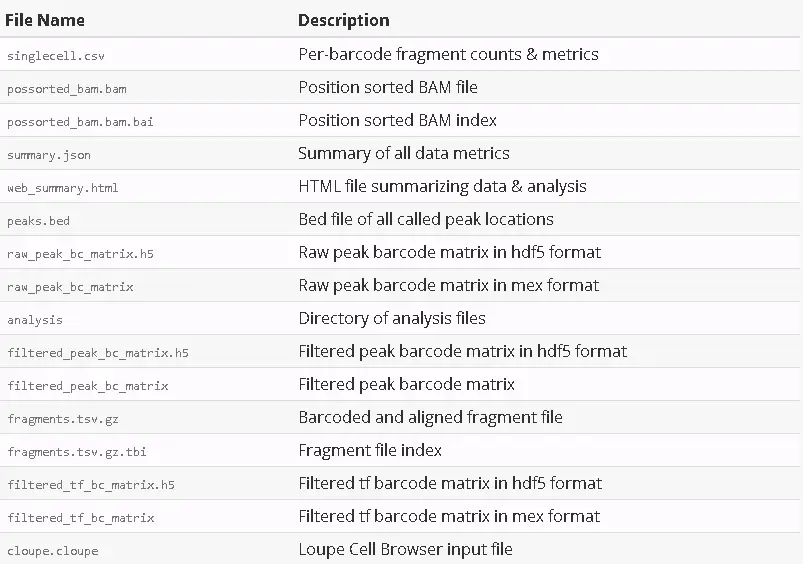
Aggregating Multiple GEM Groups with cellranger-atac aggr
当我们使用多个GEM wells构建不同的测序文库时,首先对每个GEM wells的测序数据单独进行cellranger-atac count处理,然后再使用cellranger-atac aggr子程序将不同测序文库的结果进行整合。
cellranger-atac aggr子程序使用一个逗号分隔的CSV文件作为输入,里面指定着cellranger-atac count处理好的每个文库的结果文件(specifically the fragments.tsv.gz, and singlecell.csv from each run), 最后生成一个整合好的peak-barcode matrix矩阵文件。
Requirements
For example, suppose you ran three count pipelines as follows:
$ cd /opt/runs$ cellranger-atac count --id=LV123 ...... wait for pipeline to finish ...$ cellranger-atac count --id=LB456 ...... wait for pipeline to finish ...$ cellranger-atac count --id=LP789 ...... wait for pipeline to finish ...
Now you can aggregate these three runs to get an aggregated matrix and analysis. In order to do so, you need to create an Aggregation CSV.
Setting Up An Aggregation CSV
Create a CSV file with a header line containing the following columns:
library_id: Unique identifier for this input GEM well. This will be used for labeling purposes only; it doesn’t need to match any previous ID you’ve assigned to the GEM well.fragments: Path to the fragments.tsv.gz file produced by cellranger-atac count. For example, if you processed your GEM well by calling cellranger-atac count —id=ID in some directory /DIR, the fragments would be /DIR/ID/outs/fragments.tsv.gz.cells: Path to the singlecell.csv file produced by cellranger-atac count.- (Optional)
peaks: Path to the peaks.bed file produced by cellranger-atac count. - (Optional)
Additional custom columns containing library meta-data(e.g., lab or sample origin). These custom library annotations do not affect the analysis pipeline but can be visualized downstream in the Loupe Browser.
The CSV format file would look like this:
library_id,fragments,cellsLV123,/opt/runs/LV123/outs/fragments.tsv.gz,/opt/runs/LV123/outs/singlecell.csvLB456,/opt/runs/LB456/outs/fragments.tsv.gz,/opt/runs/LB456/outs/singlecell.csvLP789,/opt/runs/LP789/outs/fragments.tsv.gz,/opt/runs/LP789/outs/singlecell.csv
Command Line Interface
These are the most common command line arguments (run cellranger-atac aggr --help for a full list):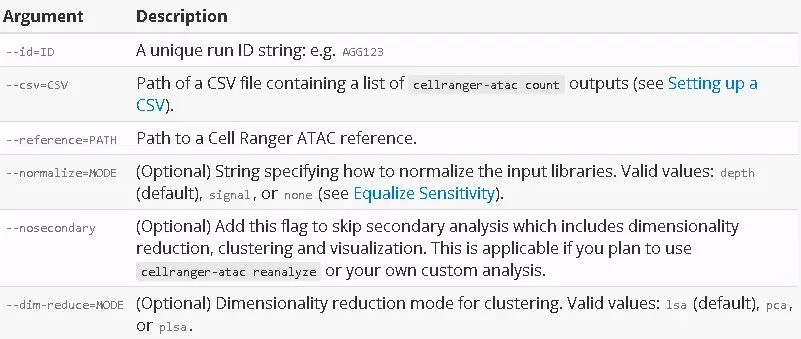
After specifying these input arguments, run cellranger-atac aggr:
$ cd /home/jdoe/runs$ cellranger-atac aggr --id=AGG123 \--csv=AGG123_libraries.csv \--normalize=depth \--reference=/home/jdoe/refs/hg19
The pipeline will begin to run, creating a new folder named with the aggregation ID you specified (e.g. /home/jdoe/runs/AGG123) for its output. If this folder already exists, cellranger-atac will assume it is an existing pipestance and attempt to resume running it.
Pipeline Outputs
The cellranger-atac aggr pipeline generates output files that contain all of the data from the individual input jobs, aggregated into single output files, for convenient multi-sample analysis. The GEM well suffix of each barcode is updated to prevent barcode collisions, as described below.
A successful run should conclude with a message similar to this:
2019-03-21 10:14:34 [runtime] (run:hydra) ID.AGG123.SC_ATAC_AGGREGATOR_CS.CLOUPE_PREPROCESS.fork0.join2019-03-21 10:14:40 [runtime] (join_complete) ID.AGG123.SC_ATAC_AGGREGATOR_CS.CLOUPE_PREPROCESS2019-03-21 10:14:40 [runtime] VDR killed 281 files, 42 MB.Outputs:- Barcoded and aligned fragment file: /home/jdoe/runs/AGG123/outs/fragments.tsv.gz- Fragment file index: /home/jdoe/runs/AGG123/outs/fragments.tsv.gz.tbi- Per-barcode fragment counts & metrics: /home/jdoe/runs/AGG123/outs/singlecell.csv- Bed file of all called peak locations: /home/jdoe/runs/AGG123/outs/peaks.bed- Filtered peak barcode matrix in hdf5 format: /home/jdoe/runs/AGG123/outs/filtered_peak_bc_matrix.h5- Filtered peak barcode matrix in mex format: /home/jdoe/runs/AGG123/outs/filtered_peak_bc_matrix- Directory of analysis files: /home/jdoe/runs/AGG123/outs/analysis- HTML file summarizing aggregation analysis : /home/jdoe/runs/AGG123/outs/web_summary.html- Filtered tf barcode matrix in hdf5 format: /home/jdoe/runs/AGG123/outs/filtered_tf_bc_matrix.h5- Filtered tf barcode matrix in mex format: /home/jdoe/runs/AGG123/outs/filtered_tf_bc_matrix- Loupe Cell Browser input file: /home/jdoe/runs/AGG123/outs/cloupe.cloupe- csv summarizing important metrics and values: /home/jdoe/runs/AGG123/outs/summary.csv- Summary of all data metrics: /home/jdoe/runs/AGG123/outs/summary.json- Annotation of peaks with genes: /home/jdoe/runs/AGG123/outs/peak_annotation.tsv- Csv of aggregation of libraries: /home/jdoe/runs/AGG123/outs/aggregation_csv.csvPipestance completed successfully!
Once cellranger-atac aggr has successfully completed, you can browse the resulting summary HTML file in any supported web browser, open the .cloupe file in Loupe Browser.
Depth Normalization: equalize sensitivity
When combining data from multiple GEM groups, the cellranger-atac aggr pipeline automatically equalizes the sensitivity of the groups before merging, which is the recommended approach in order to avoid the batch effect introduced by sequencing depth.
There are three normalization modes:
depth: (default) Subsample fragments from higher-depth GEM wells until they all have an equal number of unique fragments per cell.none: Do not normalize at all.signal: Subsample fragments from GEM wells such that each GEM well library has the same distribution of enriched cut sites along the genome.
Customized Secondary Analysis using cellranger-atac reanalyze
The cellranger-atac reanalyze command reruns secondary analysis performed on the peak-barcode matrix (dimensionality reduction, clustering and visualization) using different parameter settings.
Command Line Interface
These are the most common command line arguments (run cellranger-atac reanalyze --help for a full list):
After specifying these input arguments, run cellranger-atac reanalyze. In this example, we’re reanalyzing the results of an aggregation named AGG123:
$ cd /home/jdoe/runs$ ls -1 AGG123/outs/*.gz # verify the input file existsAGG123/outs/fragments.tsv.gz$ cellranger-atac reanalyze --id=AGG123_reanalysis \--peaks=AGG123/outs/peaks.bed \--params=AGG123_reanalysis.csv \--reference=/home/jdoe/refs/hg19 \--fragments=/home/jdoe/runs/AGG123/outs/fragments.tsv.gz
The pipeline will begin to run, creating a new folder named with the reanalysis ID you specified (e.g. /home/jdoe/runs/AGG123_reanalysis) for its output. If this folder already exists, cellranger-atac will assume it is an existing pipestance and attempt to resume running it.
Pipeline Outputs
A successful run should conclude with a message similar to this:
2019-03-22 12:45:22 [runtime] (run:hydra) ID.AGG123_reanalysis.SC_ATAC_REANALYZER_CS.SC_ATAC_REANALYZER.CLOUPE_PREPROCESS.fork0.join2019-03-22 12:46:04 [runtime] (join_complete) ID.AGG123_reanalysis.SC_ATAC_REANALYZER_CS.SC_ATAC_REANALYZER.CLOUPE_PREPROCESS2019-03-22 12:46:04 [runtime] VDR killed 270 files, 18 MB.Outputs:- Summary of all data metrics: /home/jdoe/runs/AGG123_reanalysis/outs/summary.json- csv summarizing important metrics and values: /home/jdoe/runs/AGG123_reanalysis/outs/summary.csv- Per-barcode fragment counts & metrics: /home/jdoe/runs/AGG123_reanalysis/outs/singlecell.csv- Raw peak barcode matrix in hdf5 format: /home/jdoe/runs/AGG123_reanalysis/outs/raw_peak_bc_matrix.h5- Raw peak barcode matrix in mex format: /home/jdoe/runs/AGG123_reanalysis/outs/raw_peak_bc_matrix- Filtered peak barcode matrix in hdf5 format: /home/jdoe/runs/AGG123_reanalysis/outs/filtered_peak_bc_matrix.h5- Filtered peak barcode matrix in mex format: /home/jdoe/runs/AGG123_reanalysis/outs/filtered_peak_bc_matrix- Directory of analysis files: /home/jdoe/runs/AGG123_reanalysis/outs/analysis- HTML file summarizing aggregation analysis : /home/jdoe/runs/AGG123_reanalysis/outs/web_summary.html- Filtered tf barcode matrix in hdf5 format: /home/jdoe/runs/AGG123_reanalysis/outs/filtered_tf_bc_matrix.h5- Filtered tf barcode matrix in mex format: /home/jdoe/runs/AGG123_reanalysis/outs/filtered_tf_bc_matrix- Loupe Cell Browser input file: /home/jdoe/runs/AGG123_reanalysis/outs/cloupe.cloupe- Annotation of peaks with genes: /home/jdoe/runs/AGG123_reanalysis/outs/peak_annotation.tsv- Barcoded and aligned fragment file: /home/jdoe/runs/AGG123_reanalysis/outs/fragments.tsv.gz- Fragment file index: /home/jdoe/runs/AGG123_reanalysis/outs/fragments.tsv.gz.tbiPipestance completed successfully!
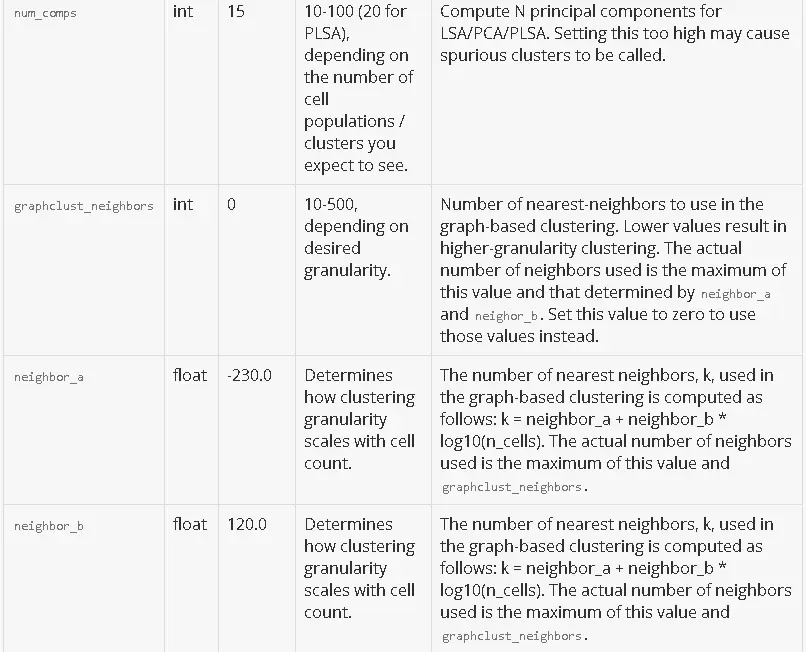
Parameters
The CSV file passed to --params should have 0 or more rows, one for every parameter that you want to customize. There is no header row. If a parameter is not specified in your CSV, its default value will be used.
Here is a detailed description of each parameter. For parameters that subset the data, a default value of null indicates that no subsetting happens by default.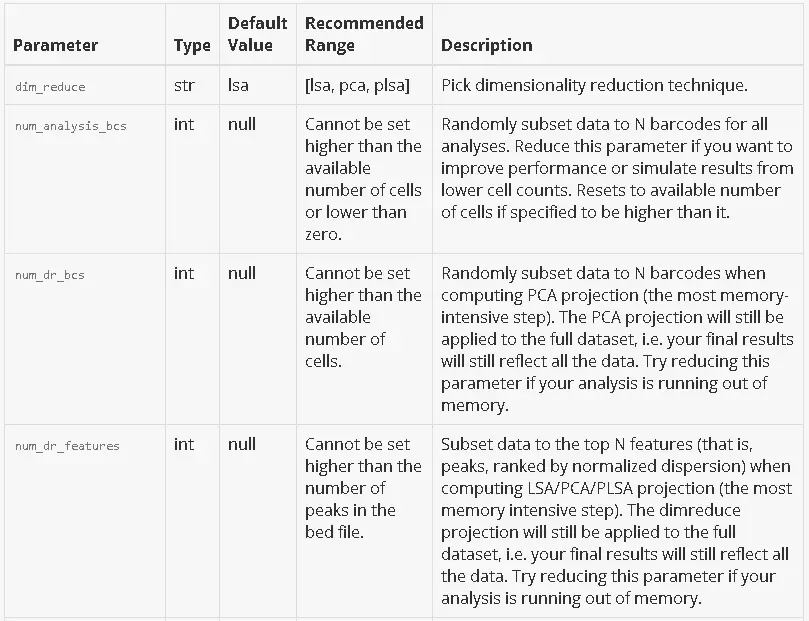
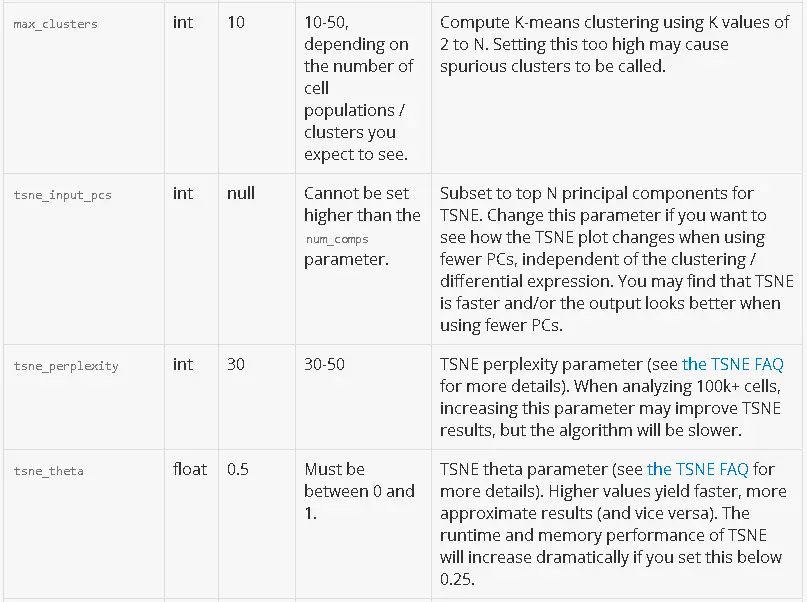
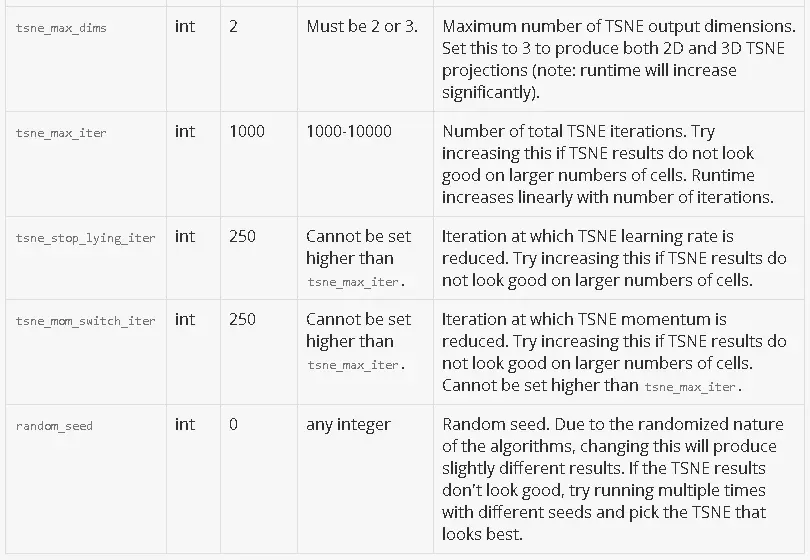
Common Use Cases
These examples illustrate what you should put in your --params CSV file in some common situations.
1)More Principal Components and Clusters
For very large / diverse cell populations, the defaults may not capture the full variation between cells. In that case, try increasing the number of principal components and / or clusters.
To run dimensionality reduction with 50 components and k-means with up to 30 clusters, put this in your CSV:
num_comps,50max_clusters,30
2)Less Memory Usage
You can limit the memory usage of the analysis by computing the LSA projection on a subset of cells and features. This is especially useful for large datasets (100k+ cells).
If you have 100k cells, it’s completely reasonable to use only 50% of them for LSA - the memory usage will be cut in half, but you’ll still be well equipped to detect rare subpopulations. Limiting the number of features will reduce memory even further.
To compute the LSA projection using 50000 cells and 3000 peaks, put this in your CSV:
num_dr_bcs,50000num_dr_features,3000
Note: Subsetting of cells is done randomly, to avoid bias. Subsetting of features is done by binning features by their mean expression across cells, then measuring the dispersion (a variance-like parameter) of each gene’s expression normalized to the other features in its bin.
参考来源:https://support.10xgenomics.com/single-cell-atac/software/pipelines/latest/using/count

若有收获,就点个赞吧
0 人点赞
