scater包提供了一系列的数据质量控制方法,可以对单细胞转录组数据进行严格的质量控制,它主要从以下三个方面进行质量控制QC:
- QC and filtering of cells(细胞水平的QC和过滤)
- QC and filtering of features (genes)(基因水平的QC和过滤)
- QC of experimental variables(实验变量的QC)
加载所需的R包和数据集
library(scater)data("sc_example_counts")data("sc_example_cell_info")example_sce <- SingleCellExperiment(assays = list(counts = sc_example_counts),colData = sc_example_cell_info)example_sce## class: SingleCellExperiment## dim: 2000 40## metadata(0):## assays(1): counts## rownames(2000): Gene_0001 Gene_0002 ... Gene_1999 Gene_2000## rowData names(0):## colnames(40): Cell_001 Cell_002 ... Cell_039 Cell_040## colData names(4): Cell Mutation_Status Cell_Cycle Treatment## reducedDimNames(0):## spikeNames(0):
计算QC metrics
scater使用calculateQCMetrics函数计算QC metrics,它可以对细胞和基因进行一系列的数据质量控制,其结果分别存储在colData和rowData中。默认情况下,calculateQCMetrics函数使用原始的count值计算这些QC metrics,也可以通过exprs_values参数进行修改。
# 使用calculateQCMetrics函数计算QC metricsexample_sce <- calculateQCMetrics(example_sce)# 查看细胞水平的QC metricscolnames(colData(example_sce))[1] "Cell" "Mutation_Status"[3] "Cell_Cycle" "Treatment"[5] "is_cell_control" "total_features_by_counts"[7] "log10_total_features_by_counts" "total_counts"[9] "log10_total_counts" "pct_counts_in_top_50_features"[11] "pct_counts_in_top_100_features" "pct_counts_in_top_200_features"[13] "pct_counts_in_top_500_features"head(colData(example_sce))DataFrame with 6 rows and 13 columnsCell Mutation_Status Cell_Cycle Treatment is_cell_control<character> <character> <character> <character> <logical>Cell_001 Cell_001 positive S treat1 FALSECell_002 Cell_002 positive G0 treat1 FALSECell_003 Cell_003 negative G1 treat1 FALSECell_004 Cell_004 negative S treat1 FALSECell_005 Cell_005 negative G1 treat2 FALSECell_006 Cell_006 negative G0 treat1 FALSEtotal_features_by_counts log10_total_features_by_counts<integer> <numeric>Cell_001 881 2.94546858513182Cell_002 624 2.79588001734408Cell_003 730 2.86391737695786Cell_004 728 2.86272752831797Cell_005 667 2.82477646247555Cell_006 646 2.8109042806687# 查看基因水平的QC metricscolnames(rowData(example_sce))[1] "is_feature_control" "mean_counts" "log10_mean_counts"[4] "n_cells_by_counts" "pct_dropout_by_counts" "total_counts"[7] "log10_total_counts"head(rowData(example_sce))DataFrame with 6 rows and 7 columnsis_feature_control mean_counts log10_mean_counts n_cells_by_counts<logical> <numeric> <numeric> <integer>Gene_0001 FALSE 252.25 2.40354945403232 17Gene_0002 FALSE 366.05 2.56472522840747 27Gene_0003 FALSE 191.65 2.28476901334902 13Gene_0004 FALSE 178.35 2.25370138101199 21Gene_0005 FALSE 0.975 0.295567099962479 13Gene_0006 FALSE 185.225 2.27003798294626 16pct_dropout_by_counts total_counts log10_total_counts<numeric> <integer> <numeric>Gene_0001 57.5 10090 4.00393420617371Gene_0002 32.5 14642 4.16563006237618Gene_0003 67.5 7666 3.88462546325623Gene_0004 47.5 7134 3.85339397745067Gene_0005 67.5 39 1.60205999132796Gene_0006 60 7409 3.86981820797933
当然,我们也可以设置一些参照(如ERCC spike-in,线粒体基因,死亡的细胞等),计算其相应的QC metrics进行质量控制。
example_sce <- calculateQCMetrics(example_sce,feature_controls = list(ERCC = 1:20, mito = 500:1000),cell_controls = list(empty = 1:5, damaged = 31:40))all_col_qc <- colnames(colData(example_sce))all_col_qc <- all_col_qc[grep("ERCC", all_col_qc)]all_col_qc[1] "total_features_by_counts_ERCC"[2] "log10_total_features_by_counts_ERCC"[3] "total_counts_ERCC"[4] "log10_total_counts_ERCC"[5] "pct_counts_ERCC"[6] "pct_counts_in_top_50_features_ERCC"[7] "pct_counts_in_top_100_features_ERCC"[8] "pct_counts_in_top_200_features_ERCC"[9] "pct_counts_in_top_500_features_ERCC"
细胞水平的QC metrics
- total_counts: total number of counts for the cell (i.e., the library size).
- total_features_by_counts: the number of features for the cell that have counts above the detection limit (default of zero).
- pct_counts_X: percentage of all counts that come from the feature control set named X.
基因水平的QC metrics
- mean_counts: the mean count of the gene/feature.
- pct_dropout_by_counts: the percentage of cells with counts of zero for each gene.
- pct_counts_Y: percentage of all counts that come from the cell control set named Y.
QC结果的可视化
Examining the most expressed features
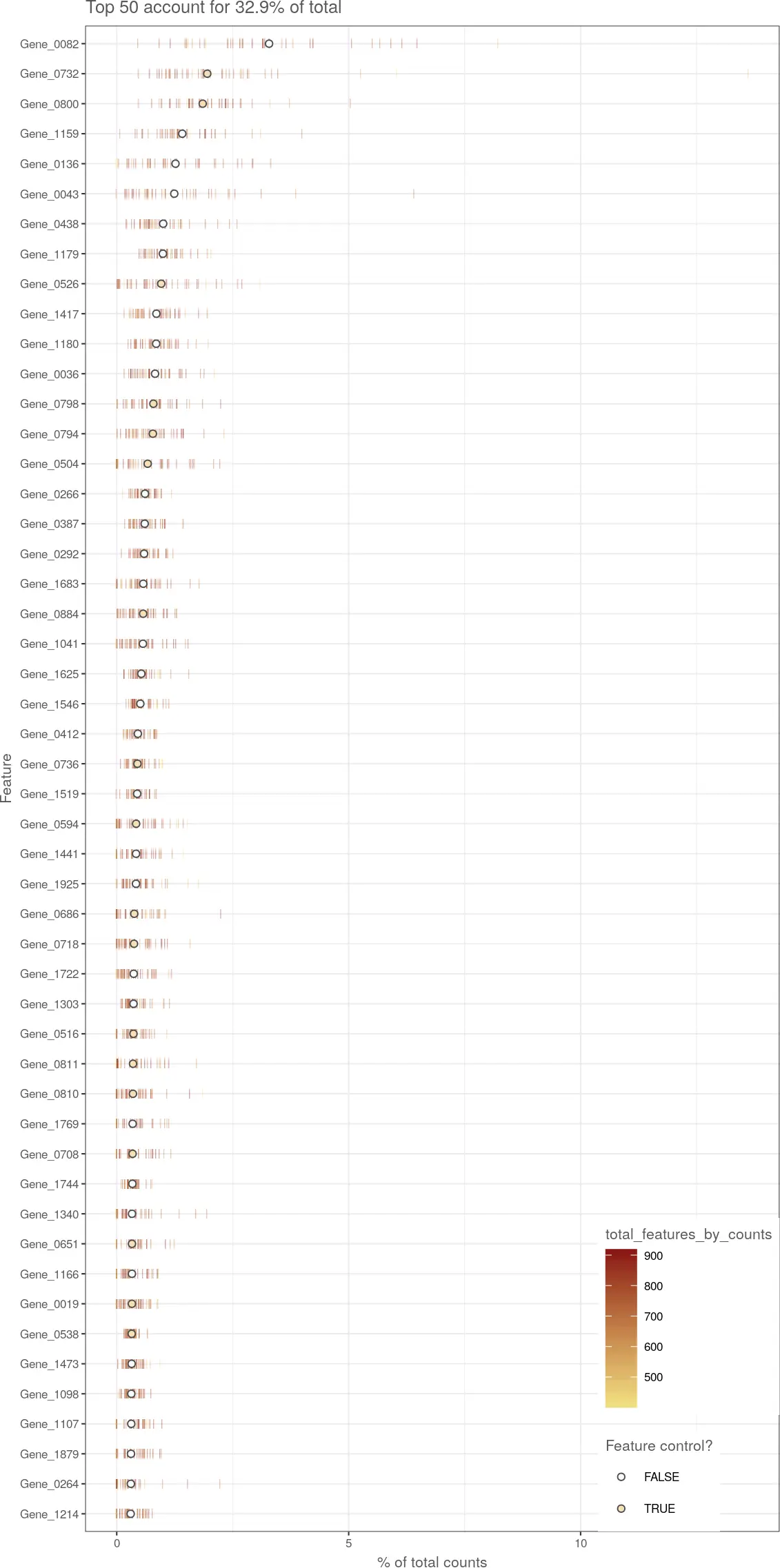
使用plotHighestExprs函数可视化那些高表达基因(默认查看50个基因)的表达情况。下图中行表示每个基因,橙色的线(bar)代表该基因在每一个细胞中的表达量,圆圈代表这个基因在所有细胞中表达量的中位数。默认情况下,使用基因的count值计算表达情况,也可以使用exprs_values参数进行修改。
plotHighestExprs(example_sce, exprs_values = "counts")
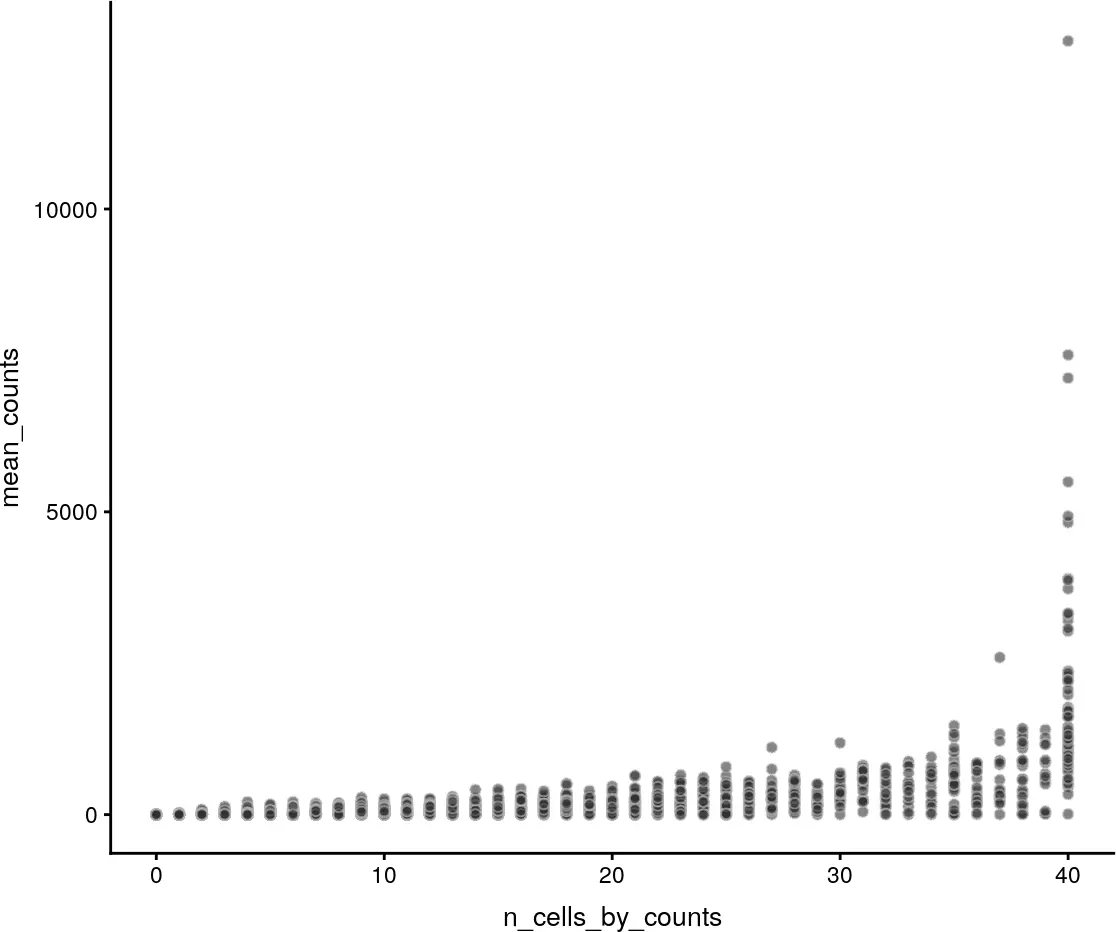
Frequency of expression as a function of the mean
使用plotExprsFreqVsMean函数进行可视化
plotExprsFreqVsMean(example_sce)
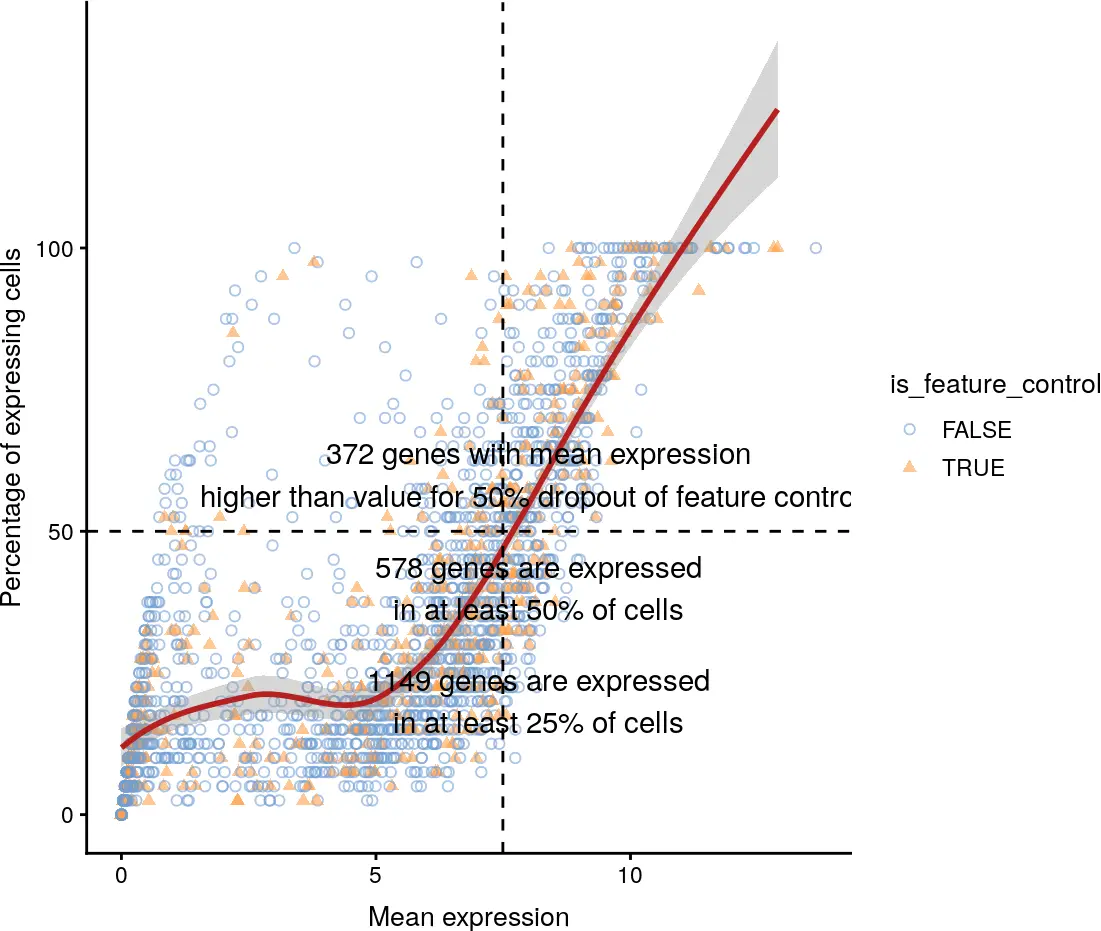
上图趋势中的异常值可能需要进一步的调查。例如,高表达基因的pseudo-genes的比对错误将导致均值低的基因在所有的细胞中表达。相反,PCR的扩增偏差(或稀有种群的存在)可能会导致在极少数细胞中表达具有很高均值的基因。
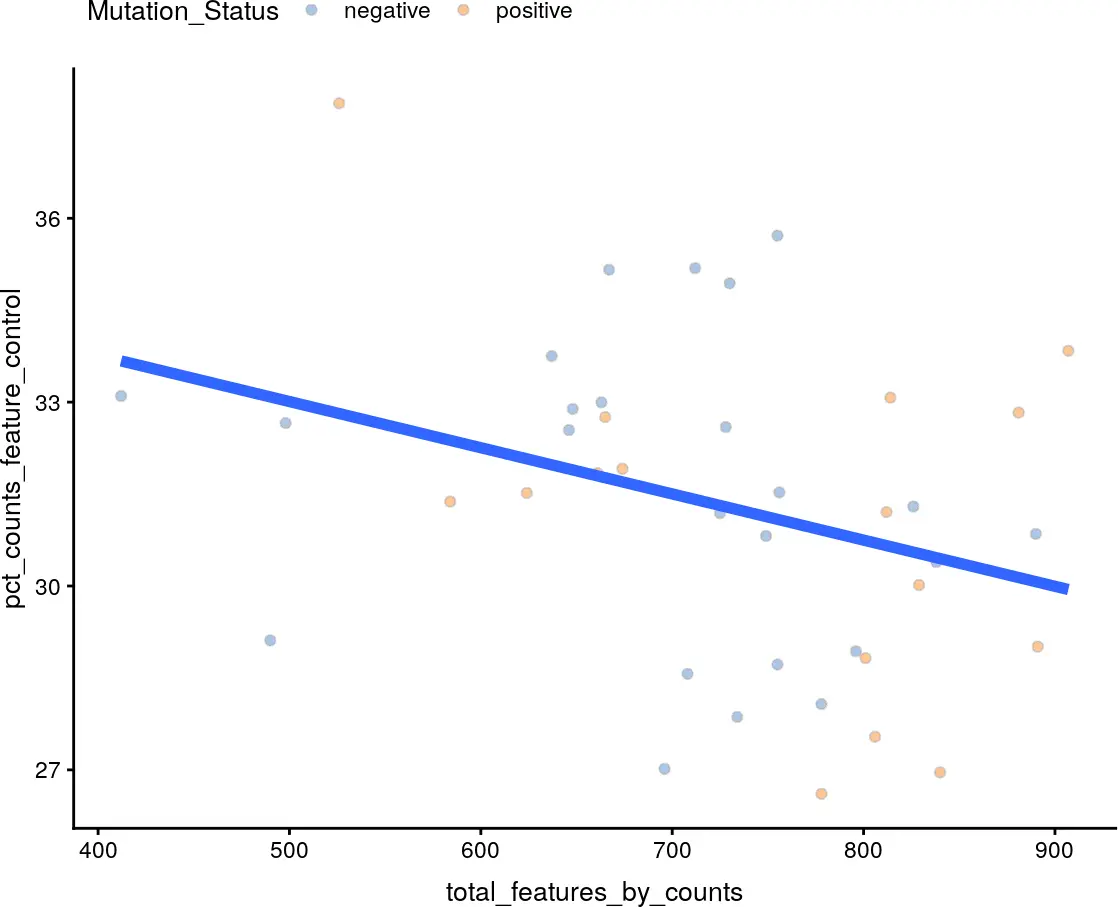
Percentage of counts assigned to feature controls
对于细胞水平上的质控,我们可以查看参照基因(feature controls)的表达量比上总基因表达量的百分比,如果一个基因在总基因表达量上的比例多,而在参照基因(如ERCC)里少,就是正常的细胞,反之则不正常。
plotColData(example_sce, x = "total_features_by_counts",y = "pct_counts_feature_control", colour = "Mutation_Status") +theme(legend.position = "top") +stat_smooth(method = "lm", se = FALSE, size = 2, fullrange = TRUE)
Cumulative expression plot
plotScater函数会从表达量最高的基因(默认为500个)中选一部分,然后从高到低累加,看看它们对每个细胞文库的贡献值大小。这种类型的图类似于对芯片数据或bulk RNA-seq数据中按样本绘制箱线图可视化不同样本的表达分布差异。累积表达图更适用于单细胞数据,因为单细胞数据难以一次性查看所有细胞的表达分布的箱形图。
为了查看不同细胞的表达分布差异,我们可以利用colData中的变量将细胞进行分类。默认使用counts值进行绘图,我们也可以通过exprs_values参数指定其他的数据。
plotScater(example_sce, block1 = "Mutation_Status", block2 = "Treatment",colour_by = "Cell_Cycle", nfeatures = 300, exprs_values = "counts")
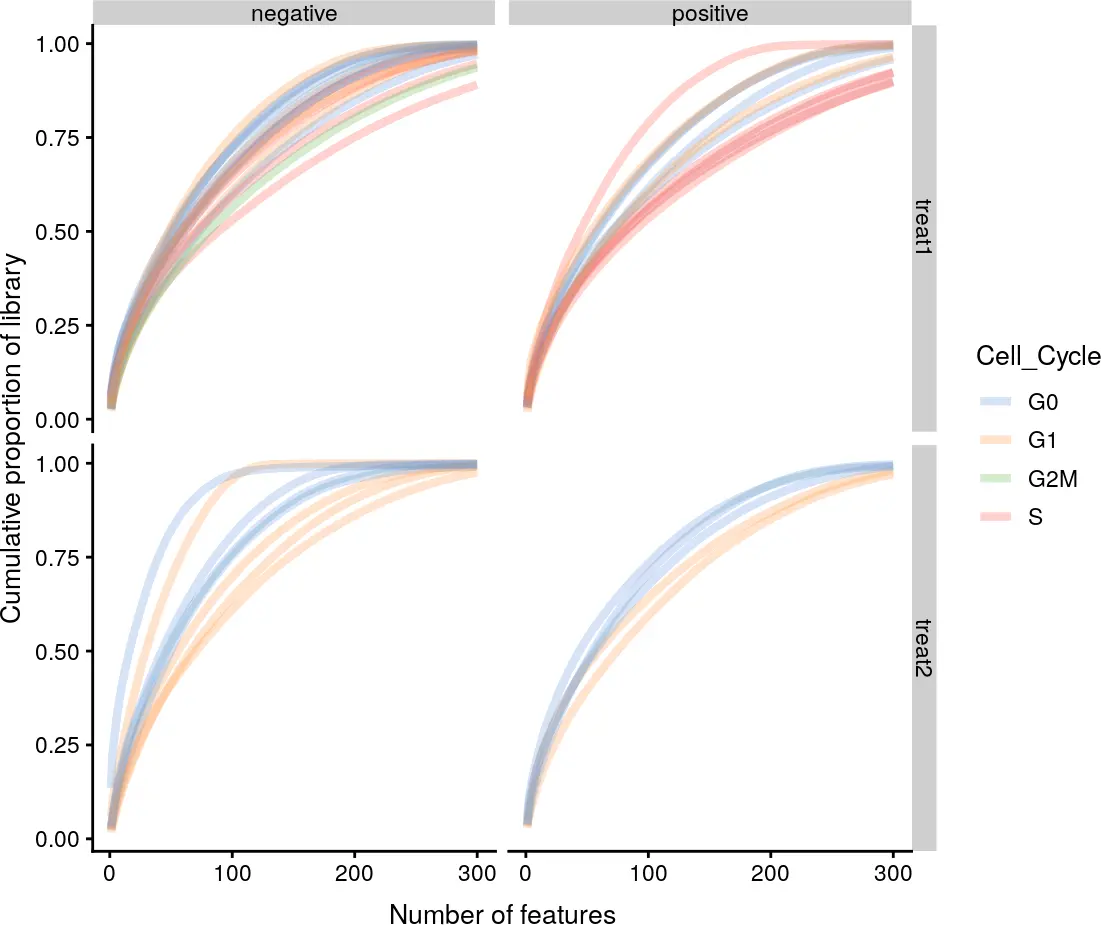
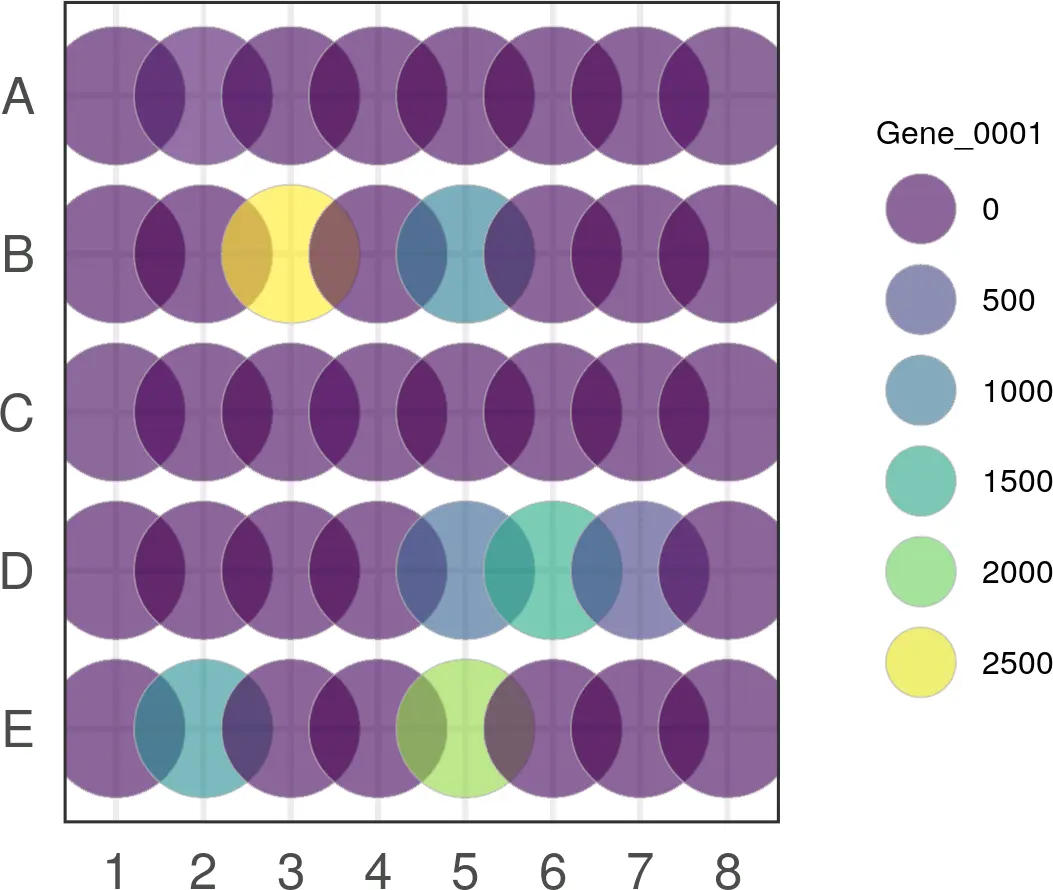
Plate position plot
For plate-based experiments, it is useful to see how expression or factors vary with the position of cell on the plate. This can be visualized using the plotPlatePosition function:
example_sce2 <- example_sceexample_sce2$plate_position <- paste0(rep(LETTERS[1:5], each = 8),rep(formatC(1:8, width = 2, flag = "0"), 5))plotPlatePosition(example_sce2, colour_by = "Gene_0001",by_exprs_values = "counts")
Other quality control plots
可以使用plotFeatureData函数轻松地查看任意两个元数据变量之间的关系:
plotRowData(example_sce, x = "n_cells_by_counts", y = "mean_counts")
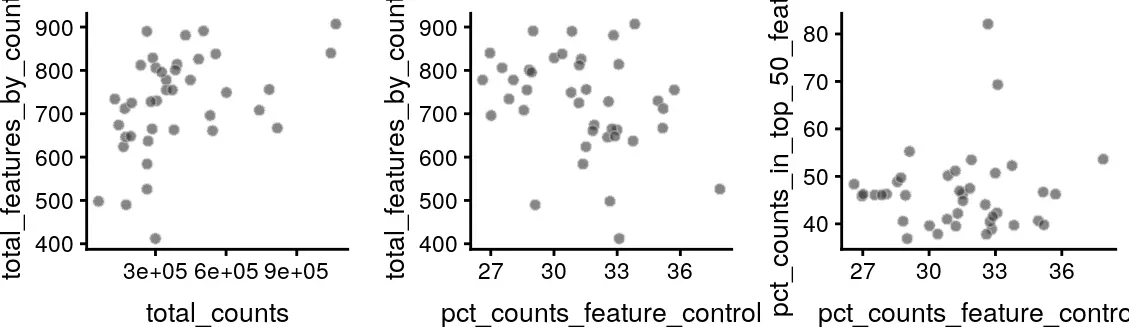
The multiplot function also allows multiple plots to be generated on the same page, as demonstrated below.
p1 <- plotColData(example_sce, x = "total_counts",y = "total_features_by_counts")p2 <- plotColData(example_sce, x = "pct_counts_feature_control",y = "total_features_by_counts")p3 <- plotColData(example_sce, x = "pct_counts_feature_control",y = "pct_counts_in_top_50_features")multiplot(p1, p2, p3, cols = 3)
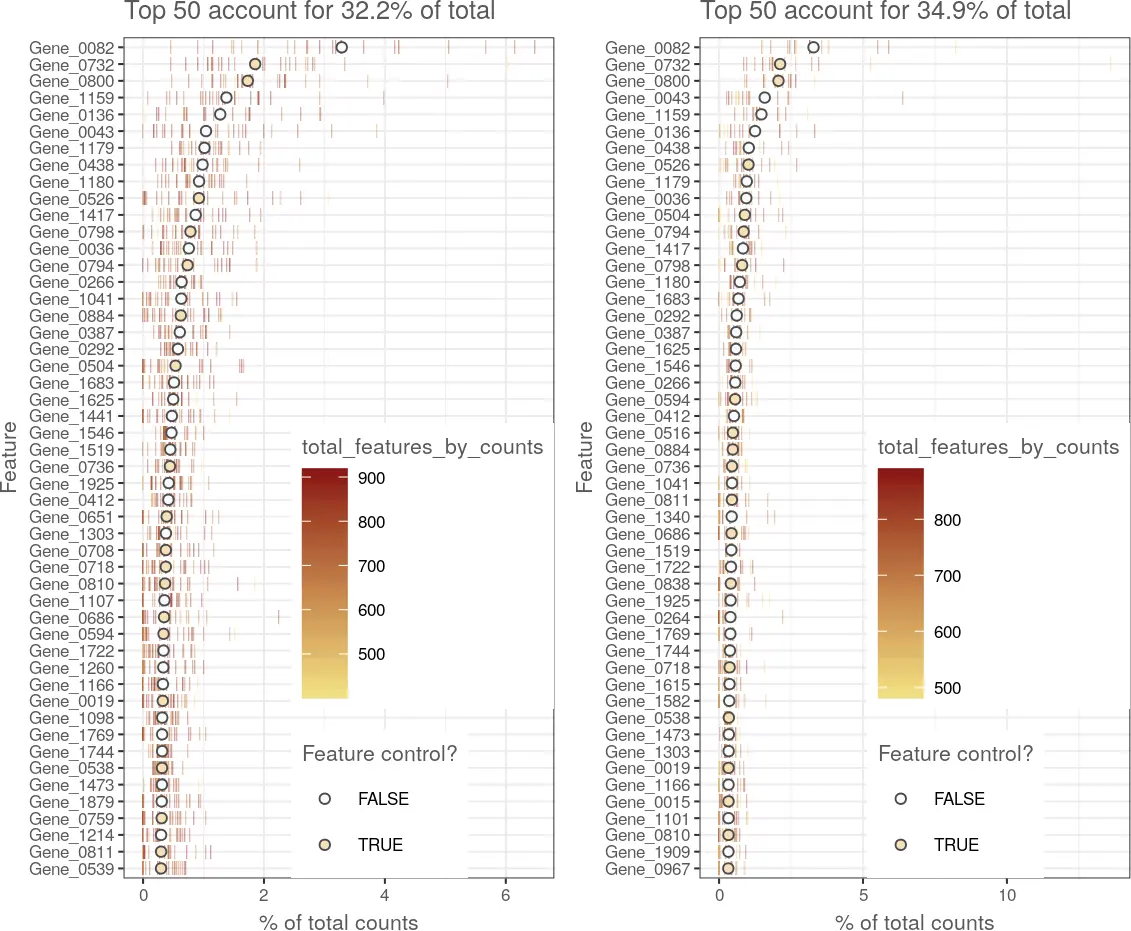
This is especially useful for side-by-side comparisons between control sets, as demonstrated below for the plot of highest-expressing features. A plot for non-control cells is shown on the left while the plot for the controls is shown on the right.
p1 <- plotHighestExprs(example_sce[, !example_sce$is_cell_control])p2 <- plotHighestExprs(example_sce[, example_sce$is_cell_control])multiplot(p1, p2, cols = 2)
QC结果的过滤
细胞水平的过滤
直接通过列数选取想要的细胞
# 选取前40个细胞example_sce <- example_sce[,1:40]
使用filter函数根据指定条件选取想要的细胞
filter(example_sce, Treatment == "treat1")## class: SingleCellExperiment## dim: 2000 27## metadata(0):## assays(1): counts## rownames(2000): Gene_0001 Gene_0002 ... Gene_1999 Gene_2000## rowData names(37): is_feature_control is_feature_control_ERCC ...## log10_total_counts_damaged pct_counts_damaged## colnames(27): Cell_001 Cell_002 ... Cell_037 Cell_039## colData names(51): Cell Mutation_Status ...## pct_counts_in_top_200_features_mito## pct_counts_in_top_500_features_mito## reducedDimNames(0):## spikeNames(0):
根据QC metrics设定阈值筛选高质量的细胞,这里我们选取那些总counts数大于100,000,表达的基因数大于500的细胞。
# 选取总counts数大于100,000的keep.total <- example_sce$total_counts > 1e5# 选取表达的基因数大于500的keep.n <- example_sce$total_features_by_counts > 500# 根据设定的条件进行过滤filtered <- example_sce[,keep.total & keep.n]dim(filtered)## [1] 2000 37
我们还可以通过isOutlier函数计算筛选的阈值,它将阈值定义为距离中位数一定数量的“中位数绝对偏差(MAD)”。超出此阈值的值被认为是异常值,可以假定它们是一些低质量的细胞,而将其过滤掉。这里我们选取那些log(total counts)值小于3倍MAD值的细胞作为outliers。
keep.total <- isOutlier(example_sce$total_counts, nmads=3,type="lower", log=TRUE)filtered <- example_sce[,keep.total]
基因水平的过滤
直接通过基因的表达量过滤掉那些低表达的基因,这里我们选取那些至少在4个细胞中表达的基因。
keep_feature <- nexprs(example_sce, byrow=TRUE) >= 4example_sce <- example_sce[keep_feature,]dim(example_sce)## [1] 1753 40
当然,我们也可以通过一些其他的条件(如核糖体蛋白基因,线粒体基因等)进行基因的过滤。
Relationships between experimental factors and expression
我们可以使用plotExplanatoryVariables函数查看不同解释因素的相对重要性。当对每个基因的不同因子进行表达量的线性回归模型拟合时,我们会对colData(example_sce)中的每个因子计算其对应的R2值。最好在表达量的对数值上执行此操作,以减少平均值对方差的影响。因此,我们首先对基因的表达量进行归一化处理。
# 先对基因的表达进行归一化处理example_sce <- normalize(example_sce)plotExplanatoryVariables(example_sce)
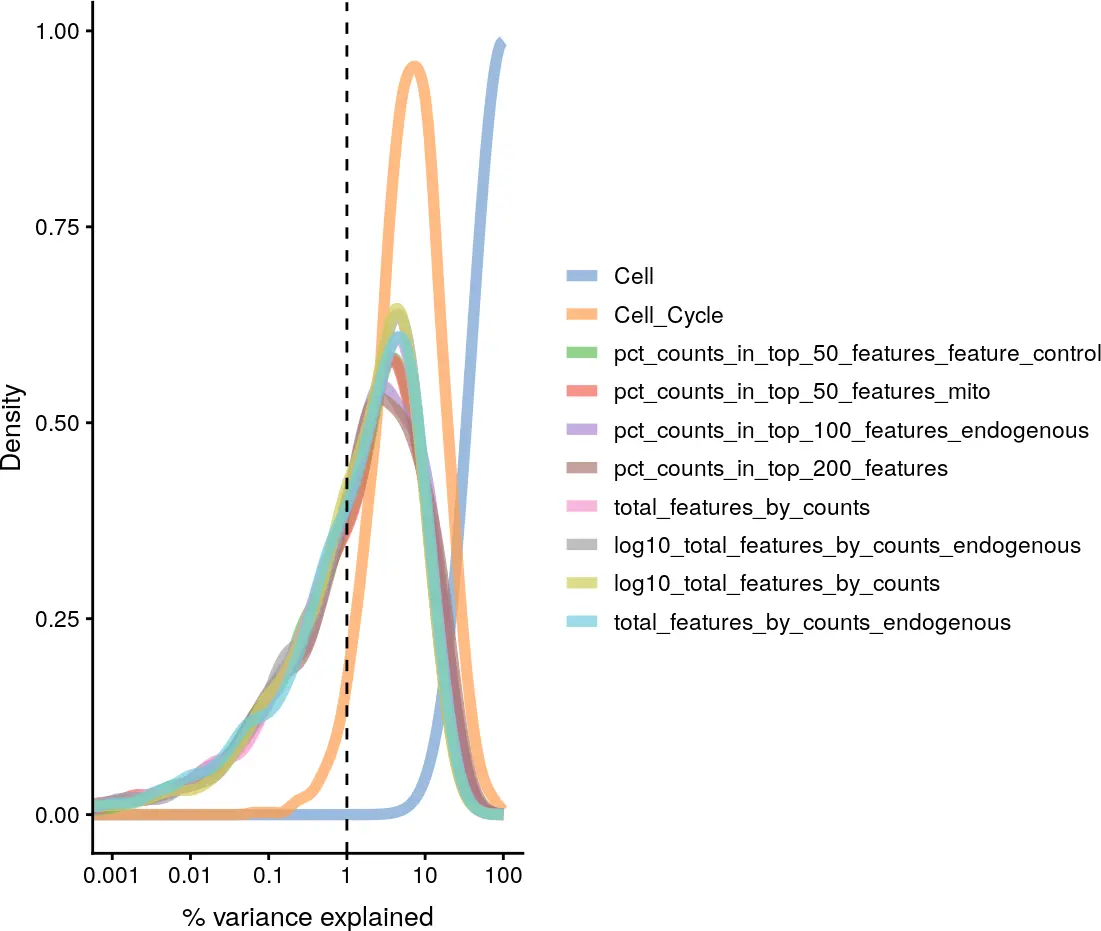
上图中每条线对应一个因子,代表所有基因中R2值的分布。当然,我们也可以通过variables参数选择特定的因子进行计算可视化。
plotExplanatoryVariables(example_sce,variables = c("total_features_by_counts", "total_counts","Mutation_Status", "Treatment", "Cell_Cycle"))
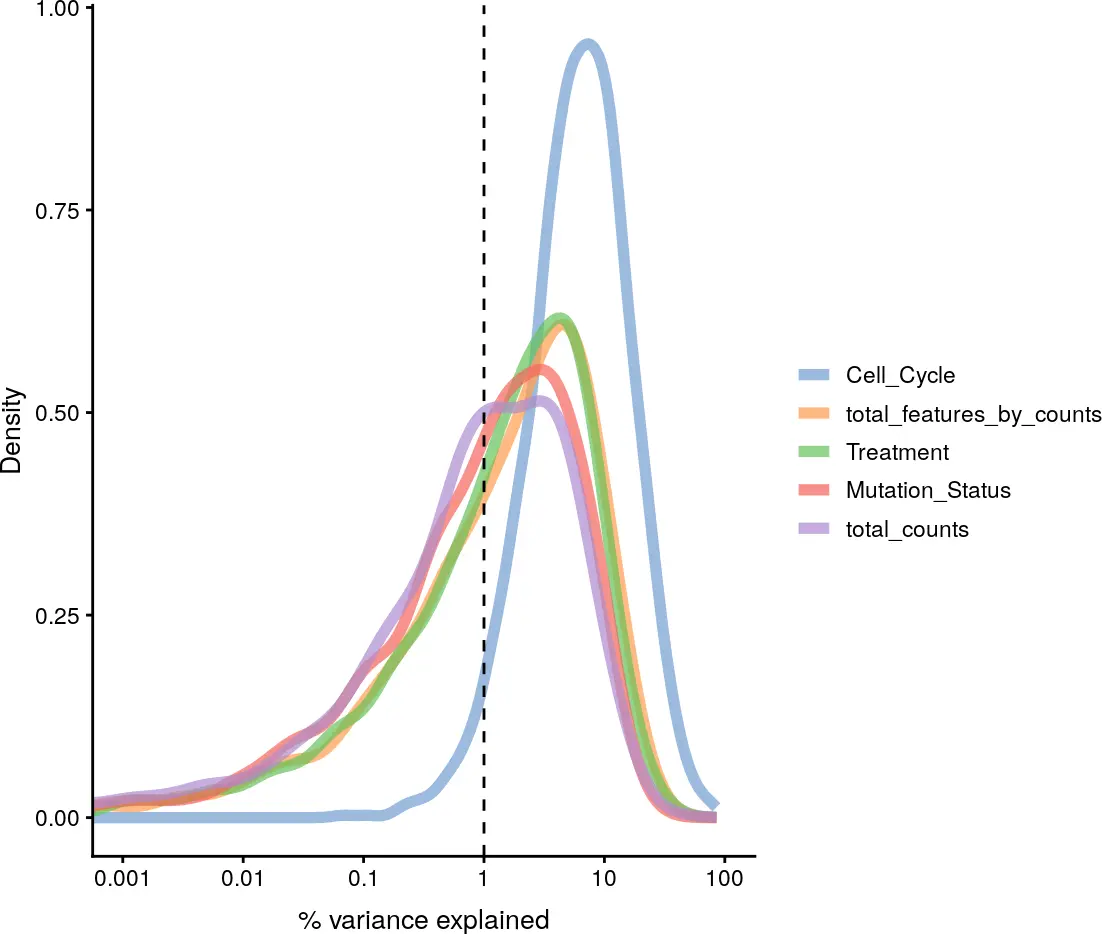
在这个小数据集中,total_counts和total_features_by_counts解释了基因表达中很大一部分的方差,它们在真实数据集中能解释的方差比例应该小得多(例如1-5%)。
Removing technical biases 去除技术偏差
Scaling normalization 数据归一化处理
缩放归一化(Scaling normalization)可以消除细胞特异性偏差,其使特定细胞中所有基因的表达增加或减少,例如测序的覆盖率或捕获效率。
进行缩放归一化的最简便方法是根据所有细胞的缩放文库大小定义size factors,使得平均size factor等于1,确保归一化后的值与原始count值的范围相同。
# 使用librarySizeFactors函数计算细胞文库size factorssizeFactors(example_sce) <- librarySizeFactors(example_sce)summary(sizeFactors(example_sce))## Min. 1st Qu. Median Mean 3rd Qu. Max.## 0.1463 0.6609 0.8112 1.0000 1.2533 2.7356
然后再使用normalize函数计算log转换后的归一化值,并将其存储在“logcounts” Assay中
example_sce <- normalize(example_sce)
虽然这种归一化的方式很简单,但细胞文库大小归一化并不能解决高通量测序数据中经常出现的成分偏差,它也不能解释影响spike-in转录本产生的差异。我们强烈建议使用来自scran包的computeSumFactors和computeSpikeFactors函数来进行计算。
Batch correction 校正批次效应
批次效应的校正可以解决不同批次中细胞之间表达的系统差异,与比例偏差不同,这些偏差通常在给定批次的所有细胞中都是恒定的,但对于每个基因而言都是不同的。
我们可以使用limma软件包中的removeBatchEffect函数来消除批次效应。
Rlibrary(limma)batch <- rep(1:2, each=20)# 使用removeBatchEffect函数去除批次效应corrected <- removeBatchEffect(logcounts(example_sce), block=batch)assay(example_sce, "corrected_logcounts") <- corrected
参考来源:http://www.bioconductor.org/packages/release/bioc/vignettes/scater/inst/doc/overview.html



若有收获,就点个赞吧
0 人点赞
