
cellassign包简介
cellassign基于marker基因的信息自动将单细胞RNA-seq数据分配注释到已知的细胞类型中。它以marker基因的细胞类型矩阵作为输入,提供先验的已知marker基因是否属于某种细胞类型。然后,cellassign会概率性地将每个细胞分配给一个细胞类型,从而消除了典型无监督聚类中的主观偏见。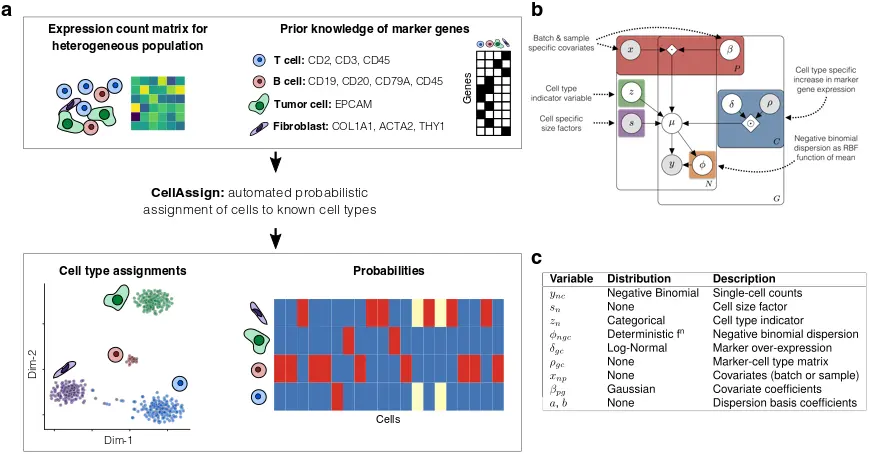
cellassign包的安装
Installing from GitHub
cellassign is built using Google's Tensorflow, and as such requires installation of the R package tensorflow:
# 安装tensorflow包install.packages("tensorflow")tensorflow::install_tensorflow(extra_packages='tensorflow-probability', version = "2.1.0")# Please ensure this installs version 2 of tensorflow. You can check this by callingtensorflow::tf_config()# TensorFlow v2.1.0 (/usr/local/lib/python3.7/site-packages/tensorflow)# cellassign can then be installed from github:# 使用devtools安装cellassign包install.packages("devtools") # If not already installeddevtools::install_github("Irrationone/cellassign")
Installing from conda
With conda, install the current release version of cellassign as follows:
# 使用conda安装cellassign包
conda install -c conda-forge -c bioconda r-cellassign
基础用法
cellassign需要以下输入文件:
exprs_obj(基因表达矩阵): Cell-by-gene matrix of raw counts (or SingleCellExperiment with counts assay)marker_gene_info(标记基因列表): Binary gene-by-celltype marker gene matrix or list relating cell types to marker geness: Size factorsX: Design matrix for any patient/batch specific effects
The model can be run as follows:
cas <- cellassign(exprs_obj = gene_expression_data,
marker_gene_info = marker_gene_info,
s = s,
X = X)
# An example set of markers for the human tumour microenvironment can be loaded by calling
data(example_TME_markers)
常用函数

数据准备
下面,我们通过差异表达分析获得marker标记基因。
我们从Holik等人的bulk RNA-seq数据(Nucleic Acids Research 2017)中获得了3种不同细胞系的marker标记基因,数据被打包到cellassign中的holik_data中:
# 加载cellassign包
library(cellassign)
# 加载holik_data数据
data(holik_data)
# which contains a matrix of counts, where each row is a gene (index by entrez ID) and each column is a sample:
# 查看holik_data数据
head(holik_data$counts[,1:2])
#> RSCE_10_BC2CTUACXX_AGTTCC_L006_R1.bam RSCE_12_BC2CTUACXX_TGACCA_L005_R1.bam
#> 1 13 12
#> 2 0 0
#> 9 346 286
#> 10 0 1
#> 12 10 17
#> 13 1 0
# as well as a vector with the cell line of origin for each sample:
# 查看细胞系类型
head(holik_data$cell_line)
#> [1] "HCC827" "HCC827" "H2228" "H2228" "H838" "H838"
We first provide a map from entrez IDs to gene symbols:
# 转换ENTREZ基因id类型为gene symbols
library(org.Hs.eg.db)
entrez_map <- select(org.Hs.eg.db,
as.character(rownames(holik_data$counts)),
c("SYMBOL"), "ENTREZID")
#> 'select()' returned 1:1 mapping between keys and columns
gene_annotations <- entrez_map %>%
dplyr::rename(GeneID=ENTREZID,
Symbol=SYMBOL)
Then construct the DGEList object for input to limma voom, filtering out lowly expressed genes:
library(limma)
# 构建DGEList对象
dge <- DGEList(counts = holik_data$counts,
group = holik_data$cell_line,
genes = gene_annotations,
remove.zeros = TRUE)
#> Removing 3620 rows with all zero counts
# 过滤低表达的基因
genes_to_keep <- rowSums(cpm(dge$counts) > 0.5) >= 2
dge_filt <- dge[genes_to_keep,]
and finally calculate the normalization factors:
# 计算归一化因子
dge_filt <- calcNormFactors(dge_filt, method="TMM")
进行差异表达分析
接下来,我们使用limma包的voom方法对三个不同的细胞样本(HCC827, H2228, H1975)进行差异分析:
# 提取数据子集
dge_subset <- dge_filt[,dge_filt$samples$group %in% c("HCC827", "H2228", "H1975")]
# 设计差异分析建模矩阵
design <- model.matrix(~ 0+dge_subset$samples$group)
colnames(design) <- levels(dge_subset$samples$group)
# 使用voom函数进行差异分析
v <- voom(dge_subset, design)
fit <- lmFit(v, design)
Next, fit constrasts to find differentially expressed genes between cell types:
# 构建比较矩阵
contrast.matrix <- makeContrasts(H2228 - H1975,
HCC827 - H1975,
HCC827 - H2228,
levels = design)
fit2 <- contrasts.fit(fit, contrast.matrix)
fit2 <- eBayes(fit2)
Finally, compute gene summary statistics and filter to only significantly differentially expressed geens (FDR < 0.05):
# 提取显著的差异表达基因
tt <- topTable(fit2, n=Inf)
tt_sig <- tt %>%
dplyr::filter(adj.P.Val < 0.05)
# 查看差异表达基因的信息
head(tt_sig)
#> GeneID Symbol H2228...H1975 HCC827...H1975 HCC827...H2228 AveExpr
#> 1 1956 EGFR 0.68777340 4.303436 3.615662 9.208711
#> 2 23480 SEC61G -0.40115418 4.414580 4.815734 7.784072
#> 3 81552 VOPP1 -0.09640406 4.347392 4.443796 6.998559
#> 4 1362 CPD 2.12265278 -2.103571 -4.226224 8.704180
#> 5 729086 LOC729086 -0.04338772 4.384976 4.428363 6.555193
#> 6 55915 LANCL2 -0.57454448 4.021963 4.596508 6.568625
#> F P.Value adj.P.Val
#> 1 2026.616 3.447052e-12 2.982899e-08
#> 2 2003.024 3.623981e-12 2.982899e-08
#> 3 1766.548 6.199752e-12 3.232724e-08
#> 4 1671.376 7.854996e-12 3.232724e-08
#> 5 1506.362 1.224645e-11 3.628466e-08
#> 6 1479.497 1.322488e-11 3.628466e-08
提取Marker标记基因
为了获得marker基因信息,我们首先使用H1975细胞系作为基准表达(baseline expression)水平来构建对数倍数变化矩阵(log fold change matrix):
lfc_table <- tt_sig[,c("H2228...H1975", "HCC827...H1975")]
lfc_table <- lfc_table %>%
dplyr::mutate(H1975=0,
H2228=H2228...H1975,
HCC827=HCC827...H1975) %>%
dplyr::select(H1975, H2228, HCC827)
rownames(lfc_table) <- tt_sig$GeneID
然后,对于每个基因,我们减去最小对数倍数变化,因为我们想知道那些相对于最小表达水平的基因过表达的基因,我们将这些基因定义为marker标记基因:
# 定义marker基因
lfc_table <- as.matrix(lfc_table)
lfc_table <- lfc_table - rowMins(lfc_table)
lfc_table <- as.data.frame(lfc_table)
现在,我们定义一个辅助函数,用于将对数倍数变化转换为二进制矩阵。这需要一个矩阵和一个阈值,将所有小于或等于阈值的值都设置为0,其他的值都设置为1:
binarize <- function(x, threshold) {
x[x <= threshold] <- -Inf
x[x > -Inf] <- 1
x[x == -Inf] <- 0
return(x)
}
接下来,我们使用定义好的辅助函数将对数倍数变化矩阵转换为二进制矩阵。实际上,我们寻找每个基因表达的最大“gap”,并指定那些表达水平高于该“gap”的基因作为该细胞类型的marker标记基因:
# Find the biggest difference
# 寻找最大差异
maxdiffs <- apply(lfc_table, 1, function(x) max(diff(sort(x))))
# 设置阈值
thres_vals <- apply(lfc_table, 1, function(x) sort(x)[which.max(diff(sort(x)))])
# 转换为二进制矩阵
expr_mat_thres <- plyr::rbind.fill(lapply(1:nrow(lfc_table), function(i) {
binarize(lfc_table[i,], thres_vals[i])
}))
rownames(expr_mat_thres) <- rownames(lfc_table)
marker_gene_mat <- expr_mat_thres[(maxdiffs >= quantile(maxdiffs, c(.99)))
& (thres_vals <= log(2)),] %>% as.matrix
Finally, we add back in gene symbols rather than entrez ids:
suppressMessages({
symbols <- plyr::mapvalues(
rownames(marker_gene_mat),
from = gene_annotations$GeneID,
to = gene_annotations$Symbol
)
})
is_na <- is.na(symbols)
marker_gene_mat <- marker_gene_mat[!is_na,]
rownames(marker_gene_mat) <- symbols[!is_na]
And there we have a marker gene matrix for our cell types(最终得到marker基因矩阵):
# 查看marker基因矩阵
head(marker_gene_mat)
#> H1975 H2228 HCC827
#> NRK 0 0 1
#> MTAP 1 0 1
#> CP 0 1 0
#> HIST1H3F 1 0 1
#> HIST1H4E 1 0 1
#> ADIRF 1 1 0
# marker标记基因可视化
library(pheatmap)
pheatmap(t(marker_gene_mat))
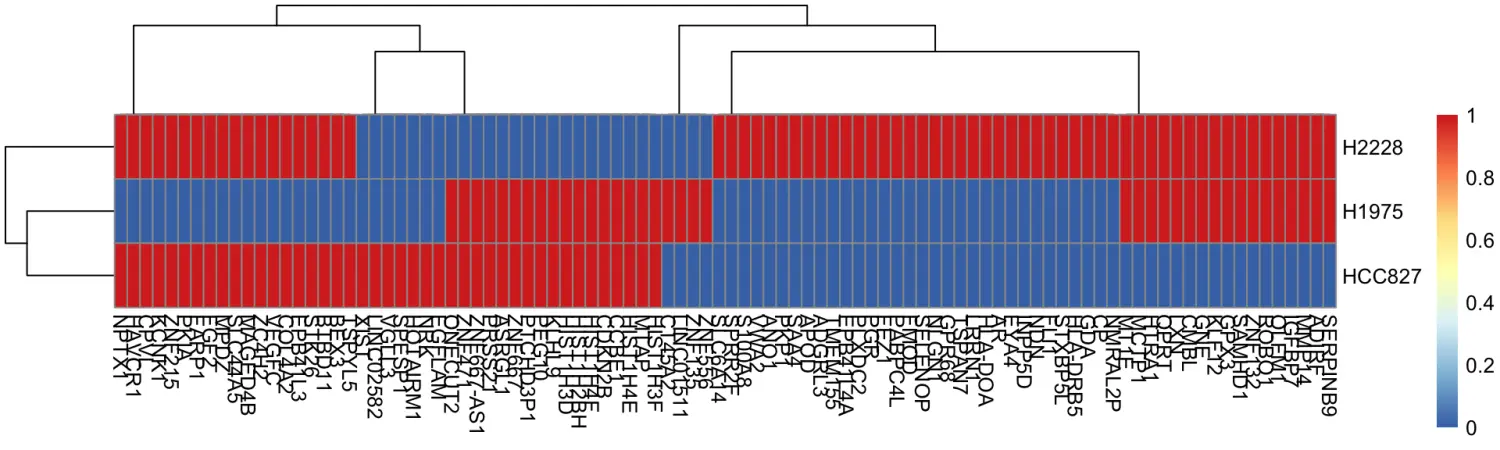
参考来源:https://irrationone.github.io/cellassign/articles/constructing-markers-from-purified-data.html

若有收获,就点个赞吧
0 人点赞
