- SnapATAC简介
- SnapATAC软件安装
- SnapATAC常见问题汇总
SnapATAC简介
SnapATAC (Single Nucleus Analysis Pipeline for ATAC-seq) 是一个能够快速、准确和全面分析单细胞ATAC-seq数据的R包,它可以对单细胞ATAC-seq数据进行常规的数据降维、聚类和批次校正分析,鉴定远端调控元件并预测其调控的靶基因,调用chromVAR软件进行motif分析,同时还可以将scRNA-seq和scATAC-seq数据进行整合分析等。
SnapATAC软件安装
Requirements
- Linux/Unix
- Python (>= 2.7 & < 3.0) (SnapTools) (highly recommanded for 2.7);
- R (>= 3.4.0 & < 3.6.0) (SnapATAC) (3.6 does not work for rhdf5 package);
Pre-print
Rongxin Fang, Sebastian Preissl, Xiaomeng Hou, Jacinta Lucero, Xinxin Wang, Amir Motamedi, Andrew K. Shiau, Eran A. Mukamel, Yanxiao Zhang, M. Margarita Behrens, Joseph Ecker, Bing Ren. Fast and Accurate Clustering of Single Cell Epigenomes Reveals Cis-Regulatory Elements in Rare Cell Types. bioRxiv 615179; doi: https://doi.org/10.1101/615179
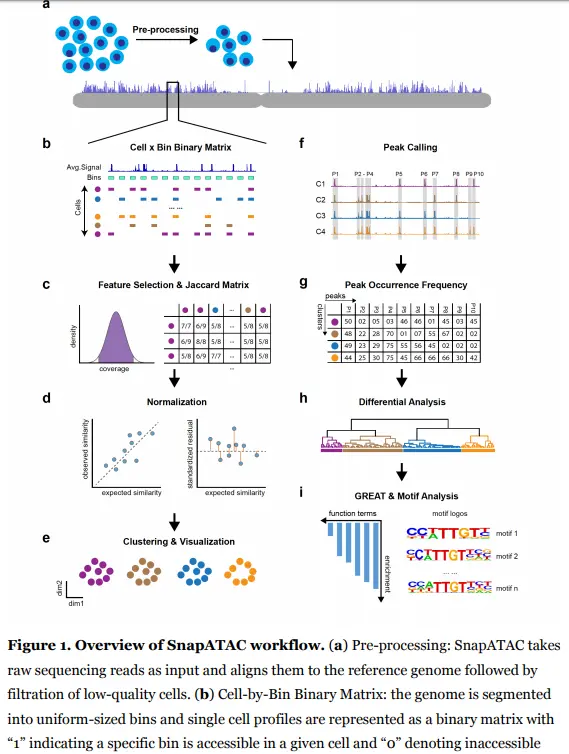
Installation
SnapATAC软件主要由以下两个组件组成:Snaptools和SnapATAC.
SnapTools- 一个用于预处理和处理snap格式文件的python模块。SnapATAC- 一个用于数据聚类、注释、motif发现和下游分析的R包。
# Install snaptools from PyPI.# NOTE: Please use python 2.7 if possible.# 使用pip安装snaptools模块pip install snaptools# Install SnapATAC R pakcage (development version).# 安装一些依赖R包install.packages(c('raster','bigmemory','doSNOW','plot3D'))# 使用devtools安装SnapATAC包library(devtools)install_github("r3fang/SnapATAC")
SnapATAC常见问题汇总
1)What is a snap file?
snap (Single-Nucleus Accessibility Profiles)格式文件是一个层级结构的hdf5文件,它可以用来存储single nucleus ATAC-seq数据集。该文件(version 4)主要由以下几个部分组成:header (HD), cell-by-bin accessibility matrix (AM), cell-by-peak matrix (PM), cell-by-gene matrix (GM), barcode (BD) and fragment (FM).
HD session:包含snap文件的版本、创建日期、比对信息和参考基因组信息。BD session:包含所有unique细胞的barcodes和相应的meta data信息。AM session:包含不同分辨率(bin size)下的cell-by-bin数据矩阵。PM session:包含cell-by-peak的计数矩阵GM session: 包含cell-by-gene的计数矩阵。FM session:包含每个细胞中可用的所有frangments片段信息。
2)How to create a snap file from fastq file?
Step 1. Barcode demultiplexing.
首先,我们将barcode信息以”@” + “barcode” + “:” + “read_name”的格式添加到每条read的开头,用以拆分FASTQ文件。
下面是一个用于拆分fastq文件的示例,我们可以通过awk或python脚本轻松实现。
# 下载示例数据wget http://renlab.sdsc.edu/r3fang/share/Fang_2019/MOs_snATAC/fastq/CEMBA180306_2B.demultiplexed.R1.fastq.gzwget http://renlab.sdsc.edu/r3fang/share/Fang_2019/MOs_snATAC/fastq/CEMBA180306_2B.demultiplexed.R2.fastq.gzwget http://renlab.sdsc.edu/r3fang/share/Fang_2019/MOs_snATAC/peaks/all_peak.bedwget http://renlab.sdsc.edu/r3fang/share/Fang_2019/MOs_snATAC/mm10.blacklist.bed.gzwget http://renlab.sdsc.edu/r3fang/share/Fang_2019/MOs_snATAC/genes/gencode.vM16.gene.bedwget http://hgdownload.cse.ucsc.edu/goldenPath/mm10/bigZips/mm10.chrom.sizeswget http://renlab.sdsc.edu/r3fang/share/Fang_2019/MOs_snATAC/genome/mm10.fa# 查看fastq文件zcat CEMBA180306_2B.demultiplexed.R1.fastq.gz | head@AGACGGAGACGAATCTAGGCTGGTTGCCTTAC:7001113:920:HJ55CBCX2:1:1108:1121:1892 1:N:0:0ATCCTGGCATGAAAGGATTTTTTTTTTAGAAAATGAAATATATTTTAAAG+DDDDDIIIIHIIGHHHIIIHIIIIIIHHIIIIIIIIIIIIIIIIIIIIII
Step 2. Index reference genome (snaptools).
接下来,我们将对参考基因组构建索引用于后续的比对分析,这里我们展示了如何使用BWA来构建参考基因组的索引。用户可以通过—-aligner参数指定要使用的比对软件,目前snaptools可以支持bwa, bowtie2和minimap2比对软件。同时,我们还需要指定比对软件所在文件夹的路径,例如,如果bwa安装在/opt/biotools/bwa/bin/bwa下,我们需要指定—path-to-aligner=/opt/biotools/bwa/bin/
# 查看bwa软件所在的路径which bwa/opt/biotools/bwa/bin/bwa# 使用snaptools构建参考基因组索引snaptools index-genome \--input-fasta=mm10.fa \--output-prefix=mm10 \--aligner=bwa \--path-to-aligner=/opt/biotools/bwa/bin/ \--num-threads=5
Step 3. Alignment (snaptools).
构建好参考基因组索引后,我们使用snaptools将拆分后的FASTQ reads序列比对到相应的参考基因组上。比对后,将比对好的bam文件按reads名称进行排序。我们还可以通过设置--num-threads参数指定多个CPU加快比对的速度。
snaptools align-paired-end \--input-reference=mm10.fa \--input-fastq1=CEMBA180306_2B.demultiplexed.R1.fastq.gz \--input-fastq2=CEMBA180306_2B.demultiplexed.R2.fastq.gz \--output-bam=CEMBA180306_2B.bam \--aligner=bwa \--path-to-aligner=/opt/biotools/bwa/bin/ \--read-fastq-command=zcat \--min-cov=0 \--num-threads=5 \--if-sort=True \--tmp-folder=./ \--overwrite=TRUE
Step 4. Pre-processing (snaptools).
比对完之后,我们将比对好的双端reads转换为fragments片段,并查看每个 fragment片段的以下属性:
1)比对质量评分MAPQ;
2)比对上的reads两端是否按比对的flag值正确配对;
3)fragments片段的长度:
我们根据以下条件进行过滤筛选,只保留满足条件的fragments片段;
1)两端正确配对的fragments片段;
2)MAPQ值大于30的fragments片段(-min-mapq);
3)长度小于1000bp的fragments片段 (-max-flen)。
# 提取参考基因染色体长度信息fetchChromSizes mm10.fa > mm10.chrom.sizes# 使用snaptools进行数据预处理,生成snap格式文件snaptools snap-pre \--input-file=CEMBA180306_2B.bam \--output-snap=CEMBA180306_2B.snap \--genome-name=mm10 \--genome-size=mm10.chrom.sizes \--min-mapq=30 \--min-flen=0 \--max-flen=1000 \--keep-chrm=TRUE \--keep-single=FALSE \--keep-secondary=FALSE \--overwrite=True \--min-cov=100 \--verbose=True
Step 5. Cell-by-bin matrix (snaptools).
最后,我们使用snap文件创建不同分辨率下的cell-by-bin矩阵文件。在下面的示例中,我们分别创建了1,000、5,000和10,000 bin size下的三个cell-by-bin矩阵,并将所有的矩阵都存储在cemba180306_2B.snap文件中。
# 使用snaptools创建cell-by-bin矩阵snaptools snap-add-bmat \--snap-file=CEMBA180306_2B.snap \--bin-size-list 1000 5000 10000 \--verbose=True
3)How to create snap file from 10X dataset?
Case 1
1)首先,运行cellranger-atac mkfastq生成拆库后的fastq文件;
2)接下来,对于每个测序文库,识别相应的测序文件,其中R1和R3是测序的reads,I1是16bp i5 (10x Barcode), R2是i7 (sample index)。
3)最后,使用snaptools提供的dex-fastq子程序,将10X barcode信息添加到reads的名称中。
snaptools dex-fastq \--input-fastq=Library1_1_L001_R1_001.fastq.gz \--output-fastq=Library1_1_L001_R1_001.dex.fastq.gz \--index-fastq-list Library1_1_L001_R2_001.fastq.gzsnaptools dex-fastq \--input-fastq=Library1_1_L001_R3_001.fastq.gz \--output-fastq=Library1_1_L001_R3_001.dex.fastq.gz \--index-fastq-list Library1_1_L001_R2_001.fastq.gzsnaptools dex-fastq \--input-fastq=Library1_2_L001_R1_001.fastq.gz \--output-fastq=Library1_2_L001_R1_001.dex.fastq.gz \--index-fastq-list Library1_2_L001_R2_001.fastq.gzsnaptools dex-fastq \--input-fastq=Library1_2_L001_R3_001.fastq.gz \--output-fastq=Library1_2_L001_R3_001.dex.fastq.gz \--index-fastq-list Library1_2_L001_R2_001.fastq.gz# combine these two librarycat Library1_1_L001_R1_001.fastq.gz Library1_2_L001_R1_001.fastq.gz > Library1_L001_R1_001.fastq.gzcat Library1_1_L001_R3_001.fastq.gz Library1_2_L001_R3_001.fastq.gz > Library1_L001_R3_001.fastq.gz
Case 2
在本示例中,我们有两个10x的测序文库(每个文库都通过单独的Chromium chip channel进行处理)。请注意,在运行完cellranger-atac mkfastq拆分数据之后,我们对每个文库进行单独的cellranger-atac count处理。
snaptools dex-fastq \--input-fastq=Library1_S1_L001_R1_001.fastq.gz \--output-fastq=Library1_S1_L001_R1_001.dex.fastq.gz \--index-fastq-list Library1_S1_L001_R2_001.fastq.gzsnaptools dex-fastq \--input-fastq=Library1_S1_L001_R3_001.fastq.gz \--output-fastq=Library1_S1_L001_R3_001.dex.fastq.gz \--index-fastq-list Library1_S1_L001_R2_001.fastq.gz
4)Can I run SnapATAC with CellRanger outcome?
Yes. There are two entry points
(1)use the position sorted bam file (recommanded).
# 查看比对的bam文件信息samtools view atac_v1_adult_brain_fresh_5k_possorted_bam.bamA00519:218:HJYFLDSXX:2:1216:26458:34976 99 chr1 3000138 60 50M = 3000474 385 TGATGACTGCCTCTATTTCTTTAGGGGAAATGGGACTTTTAGTCCATGAA FFFFFFFFFFFFFFFF,FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF NM:i:0 MD:Z:50 MC:Z:49M AS:i:50 XS:i:37 CR:Z:GGTTGCGAGCCGCAAA CY:Z:FFFFFFFFFFFFFFFF CB:Z:GGTTGCGAGCCGCAAA-1 BC:Z:AACGGTCA QT:Z:FFFFFFFFGP:i:3000137 MP:i:3000522 MQ:i:40 RG:Z:atac_v1_adult_brain_fresh_5k:MissingLibrary:1:HJYFLDSXX:2
在比对的read名称前添加barcode信息
The cell barcode is embedded in the tag CB:Z:GGTTGCGAGCCGCAAA-1, you can modify the bam file by add the cell barcode GGTTGCGAGCCGCAAA-1 to the beginning of read
# extract the header filesamtools view atac_v1_adult_brain_fresh_5k_possorted_bam.bam -H > atac_v1_adult_brain_fresh_5k_possorted.header.sam# create a bam file with the barcode embedded into the read namecat <( cat atac_v1_adult_brain_fresh_5k_possorted.header.sam ) \<( samtools view atac_v1_adult_brain_fresh_5k_possorted_bam.bam | awk '{for (i=12; i<=NF; ++i) { if ($i ~ "^CB:Z:"){ td[substr($i,1,2)] = substr($i,6,length($i)-5); } }; printf "%s:%s\n", td["CB"], $0 }' ) \| samtools view -bS - > atac_v1_adult_brain_fresh_5k_possorted.snap.bam# 查看添加好barcode信息的bam文件samtools view atac_v1_adult_brain_fresh_5k_possorted.snap.bam | cut -f 1 | headGGTTGCGAGCCGCAAA-1:A00519:218:HJYFLDSXX:2:1216:26458:34976GGTTGCGAGCCGCAAA-1:A00519:218:HJYFLDSXX:2:2256:23194:13823GGTTGCGAGCCGCAAA-1:A00519:218:HJYFLDSXX:2:2546:5258:31955CTCAGCTAGTGTCACT-1:A00519:218:HJYFLDSXX:1:2428:8648:18349CTCAGCTAGTGTCACT-1:A00519:218:HJYFLDSXX:1:2428:8648:18349GAAGTCTGTAACACTC-1:A00519:218:HJYFLDSXX:3:2546:14968:2331GAAGTCTGTAACACTC-1:A00519:218:HJYFLDSXX:3:2546:14705:2628GGTTGCGAGCCGCAAA-1:A00519:218:HJYFLDSXX:2:1216:26458:34976GGTTGCGAGCCGCAAA-1:A00519:218:HJYFLDSXX:2:2256:23194:13823GGTTGCGAGCCGCAAA-1:A00519:218:HJYFLDSXX:2:2546:5258:31955
对bam文件按read的名称进行排序
Then sort the bam file by read name:
samtools sort -n -@ 10 -m 1G atac_v1_adult_brain_fresh_5k_possorted.snap.bam -o atac_v1_adult_brain_fresh_5k.snap.nsrt.bam
使用snaptools进行数据预处理生成snap文件
Then generate the snap file
snaptools snap-pre \--input-file=atac_v1_adult_brain_fresh_5k.snap.nsrt.bam \--output-snap=atac_v1_adult_brain_fresh_5k.snap \--genome-name=mm10 \--genome-size=mm10.chrom.sizes \--min-mapq=30 \--min-flen=50 \--max-flen=1000 \--keep-chrm=TRUE \--keep-single=FALSE \--keep-secondary=False \--overwrite=True \--max-num=20000 \--min-cov=500 \--verbose=True
删除中间文件
remove temporary files
rm atac_v1_adult_brain_fresh_5k.snap.snap.bamrm atac_v1_adult_brain_fresh_5k_possorted.header.sam
(2)use the fragment tsv file. Fragment file is already filtered, this will disable snaptools to generate quality control metrics.
# decompress the gz filegunzip atac_v1_adult_brain_fresh_5k_fragments.tsv.gz# sort the tsv file using the 4th column (barcode column)sort -k4,4 atac_v1_adult_brain_fresh_5k_fragments.tsv > atac_v1_adult_brain_fresh_5k_fragments.bed# compress the bed filegzip atac_v1_adult_brain_fresh_5k_fragments.bed# run snap files using the bed filesnaptools snap-pre \--input-file=atac_v1_adult_brain_fresh_5k_fragments.bed.gz \--output-snap=atac_v1_adult_brain_fresh_5k.snap \--genome-name=mm10 \--genome-size=mm10.chrom.sizes \--min-mapq=30 \--min-flen=50 \--max-flen=1000 \--keep-chrm=TRUE \--keep-single=FALSE \--keep-secondary=False \--overwrite=True \--max-num=20000 \--min-cov=500 \--verbose=True
5)How to create a snap file from bam or bed file?
在很多情况下,我们可以直接使用snaptools pre子程序将比对好的、按read名称进行排序的bam或bed文件作为输入,生成snap格式文件。强烈建议使用未经过滤的比对文件作为输入。
(1)对于bam文件,我们需要在read的名称前添加细胞的barcode信息,如下所示:
samtools view demo.bam|headAAACTACCAGAAAGACGCAGTT:7001113:968:HMYT2BCX2:1:1215:20520:88475 77 * 0 0 * * 0 0CTATGAGCACCGTCTCCGCCTCAGATGTGTATAAGAGACAGCAGAGTAAC @DDBAI??E?1/<DCGECEHEHHGG1@GEHIIIHGGDGE@HIHEEIIHH1 AS:i:0 XS:i:0AAACTACCAGAAAGACGCAGTT:7001113:968:HMYT2BCX2:1:1215:20520:88475 141 * 0 0 * * 0 0GGCTTGTACAGAGCAAGTGCTGAAGTCCCTTTCTGATGACGTTCAACAGC 0<000/<<1<D1CC111<<1<1<111<111<<CDCF1<1<DHH<<<<C11 AS:i:0 XS:i:0AAACTACCAGAAAGACGCAGTT:7001113:968:HMYT2BCX2:1:2201:20009:41468 77 * 0 0 * * 0 0CGGTGCCCCTGTCCTGTTCGTGCCCACCGTCTCCGCCTCAGATGTGTATA DDD@D/D<DHIHEHCCF1<<CCCGH?GHI1C1DHIII0<1D1<111<1<1 AS:i:0 XS:i:0AAACTACCAGAAAGACGCAGTT:7001113:968:HMYT2BCX2:1:2201:20009:41468 141 * 0 0 * * 0 0GAGCGAGGGCGGCAGAGGCAGGGGGAGGAGACCCGGTGGCCCGGCAGGCT 0<00</<//<//<//111000/<</</<0<1<1<//<<0<DCC/<///<D AS:i:0 XS:i:0# 使用snaptools将bam文件转换为snap格式文件snaptools snap-pre \--input-file=demo.bam \--output-snap=demo.snap \--genome-name=mm10 \--genome-size=mm10.chrom.sizes \--min-mapq=30 \--min-flen=0 \--max-flen=1000 \--keep-chrm=TRUE \--keep-single=TRUE \--keep-secondary=False \--overwrite=True \--min-cov=100 \--verbose=True
(2)对于bed格式的文件,应将barcode信息添加到第四列中,如下所示:
zcat demo.bed.gz | headchr2 74358918 74358981 AACGAGAGCTAAAGACGCAGTTchr6 134212048 134212100 AACGAGAGCTAAAGACGCAGTTchr10 93276785 93276892 AACGAGAGCTAAAGACGCAGTTchr2 128601366 128601634 AACGAGAGCTAAAGCGCATGCTchr16 62129428 62129661 AACGAGAGCTAACAACCTTCTGchr8 84946184 84946369 AACGAGAGCTAACAACCTTCTG# 使用snaptools将bed文件转换为snap格式文件snaptools snap-pre \--input-file=demo.bed.gz \--output-snap=demo.snap \--genome-name=mm10 \--genome-size=mm10.chrom.sizes \--min-mapq=30 \--min-flen=0 \--max-flen=1000 \--keep-chrm=TRUE \--keep-single=TRUE \--keep-secondary=False \--overwrite=True \--min-cov=100 \--verbose=True
6)How to group reads?
(1)Group reads from one cell ATACAGCCTCGC in snap file sample1.snap.
library(SnapATAC);snap_files = "sample1.snap";barcode_sel = "ATACAGCCTCGC";reads.gr = extractReads(barcode_sel, snap_files);
(2)Group reads from multiple barcodes in one snap file.
library(SnapATAC);barcode_sel = c("ATACAGCCTCGC", "ATACAGCCTCGG")snap_files = rep("sample1.snap", 2);reads.gr = extractReads(barcode_sel, snap_files);
(3)Group reads from multiple barcodes and multiple snap files.
library(SnapATAC);barcode_sel = rep("ATACAGCCTCGC", 2);snap_files = c("sample1.snap", "sample2.snap");reads.gr = extractReads(barcode_sel, snap_files);
7)How to analyze multiple samples together?
因为SnapATAC软件使用cell-by-bin矩阵对细胞进行聚类分群,这使他很容易将多个样本进行结合并执行比较分析。它需要将所有的样本创建相同bin size大小的cell-by-bin矩阵。在这里,我们以PBMC_5K和PBMC_10K数据为例进行分析。
$ R# 加载SnapATAC包> library(SnapATAC);# 下载示例数据> system("wget http://renlab.sdsc.edu/r3fang/share/Fang_2019/published_scATAC/atac_v1_pbmc_10k_fastqs/atac_v1_pbmc_10k.snap");> system("wget http://renlab.sdsc.edu/r3fang/share/Fang_2019/published_scATAC/atac_v1_pbmc_5k_fastqs/atac_v1_pbmc_5k.snap");> file.list = c("atac_v1_pbmc_5k.snap", "atac_v1_pbmc_10k.snap");> sample.list = c("pbmc.5k", "pbmc.10k");# 使用createSnap函数将两个数据结合在一起> x.sp = createSnap(file=file.list, sample=sample.list);
createSnap函数将创建一个snap对象,该对象中包含了每个snap文件的名称和相应的barcodes信息。
8)How to choose bin size?
SnapATAC软件是基于cell-by-bin矩阵对细胞进行聚类分群的,因此选择不同的bin size可能对细胞的聚类分群产生较大的影响。如何选择最优的bin size,这个问题没有绝对的答案。
一方面,我们发现在5kb-50kb范围内的bin size大小的改变,并没有明显地改变细胞聚类分群的结果(如下图所示)。而另一方面,我们确实注意到了一个大的bin size通常会生成相对较少的cluster。这种聚类的差异,可以使用分辨率较小的Louvain聚类算法进行弥补。
使用较大bin size的好处是可以节省一些内存,这对于一些大型数据集尤为有用。这里提供了一个bin size大小选择的主观建议。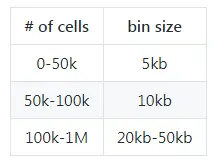
参考来源:https://github.com/r3fang/SnapATAC/wiki/FAQs#what-is-a-snap-file

若有收获,就点个赞吧
0 人点赞
