Celaref包简介
单细胞RNA-seq测序(scRNA-seq)技术可以解析组织或样品内的细胞异质性,并查看特定细胞类型的特征和变化情况。因此,在进行单细胞聚类分群后,我们需要弄清楚每个cluster簇的细胞类型。
在典型的scRNA-seq实验中,首先,我们对每个细胞的基因表达水平进行细胞计数定量。然后,根据基因表达水平的相似性对细胞进行聚类分群成不同的cluster簇。目前,有许多不同的细胞聚类工具可以做到这一点(Freytag et al. 2017)。
细胞聚类工具通常只能将相似的细胞聚成一个特定的cluster簇,但不提供有关其生物学意义的解释。对每个cluster簇的“细胞类型”的注释,主要由生物学家根据专业的生物学知识进行解析,它们通常会查看已知的marker基因的表达或差异分析以了解每个簇可能的细胞类型。
Celaref包主要通过与已知细胞类型的参考数据集的相似性进行比较,根据比较的结果对相似度高的、未知的cluster簇进行注释。Celaref需要每个细胞的基因表达矩阵,以及属于每个cluster的细胞列表(用于测试和参考数据)。它比较每个查询组中最显著富集的基因在参考样本中的排名,根据排名来匹配细胞类型。
参考文献:Sarah Williams (2019). celaref: Single-cell RNAseq cell cluster labelling by reference. R package version 1.0.1.
Celaref包工作流程
下面是典型的Celaref工作流程,它主要通过与带注释的参考数据集的转录组表达谱的相似性进行比较,来注释查询数据集中的不同clusters。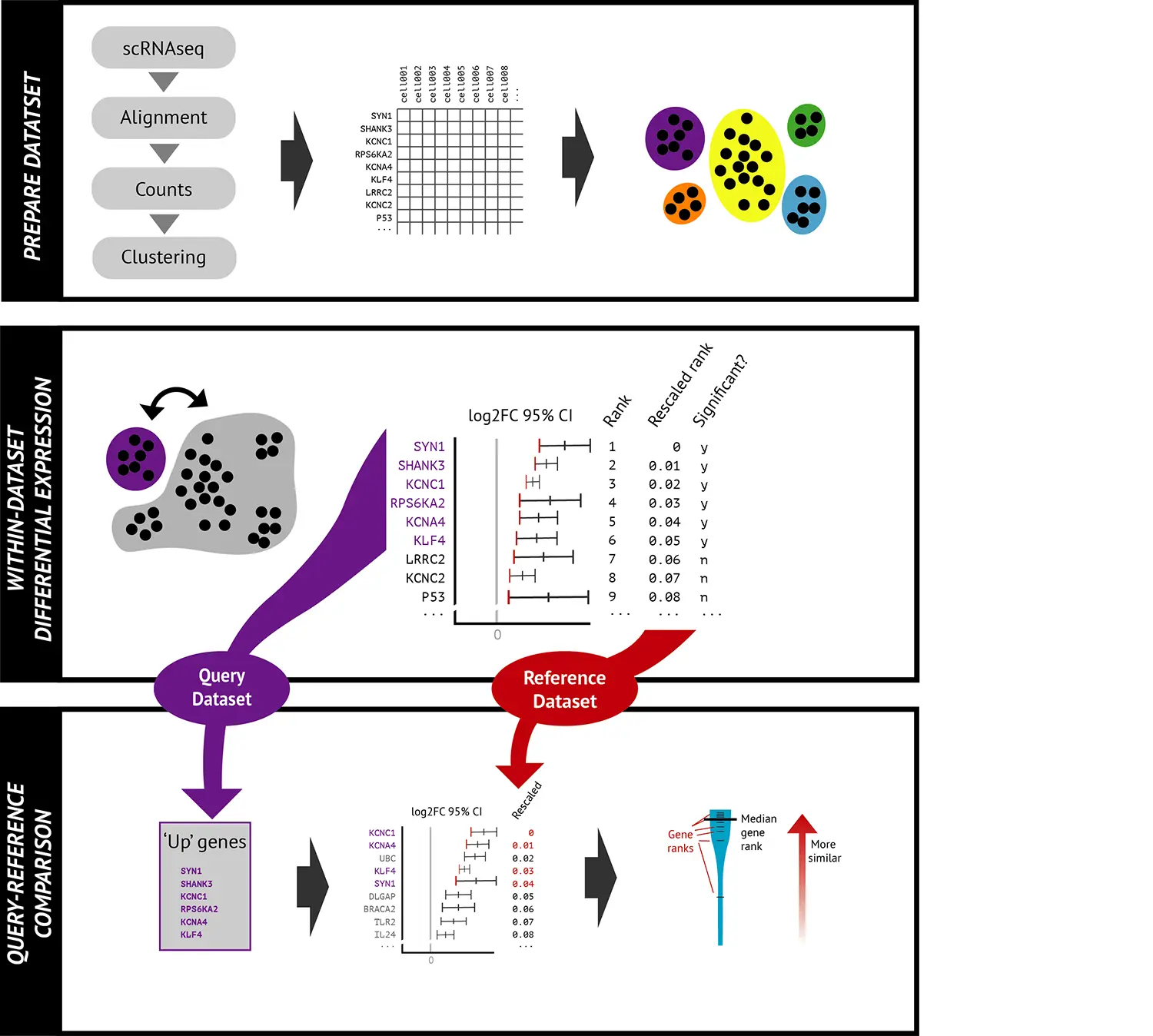
Prepare dataset 准备数据集
使用Celaref包进行细胞类型注释时,我们主要需要以下两个输入文件:
- A counts matrix of number of reads per gene, per cell(基因表达矩阵)。
gene Cell1 cell2 cell3 cell4 … cell954GeneA 0 1 0 1 … 0GeneB 0 3 0 2 … 2GeneC 1 40 1 0 … 0
- Cluster assignment for each cell(细胞分群信息).
CellId Cluster
cell1 cluster1
cell2 cluster7
… …
cell954 cluster8
Within dataset differential expression 进行差异表达分析
我们采用相同的方式准备好每个数据集(无论是查询还是参考数据集)。对于每个cluster簇,将该簇内的细胞与其他簇合并在一起的细胞进行比较,使用MAST方法进行基因的差异表达分析(Finak等人,2015)。由于单细胞RNA-seq数据中很多基因的表达较低,并存在潜在的缺失(drop-out)问题,因此仅考虑每个cluster簇中显著富集的基因。对于每个cluster簇,Celaref将细胞根据其表达水平的差异倍数的低于95%CI值从高到低排序。每个基因都有一个从0(最富集)到1(最不存在)的“rescaled rank”。此步骤耗时最久,但每个数据集只需要执行一次。
Query-Reference comparison 比较查询-参考数据集
进行差异分析后,我们可以得到每个查询细胞簇(query cluster)的“Up”基因列表,这些基因在该cluster中的表达明显高于其在其他簇中的表达(BH多重假设校正后,p<0.01)。然后,在每个参考数据集的细胞簇中查找这些基因的排名。最后,根据这些“Up“基因排名的分布,来评估查询细胞组与参考细胞组的相似性。
输出结果
Interpreting output 解释输出结果
通常,我们将查询数据集中的每个cluster簇与参考数据集(X轴)中的所有cluster簇进行作图。其中,刻度线表示up基因,中间基因等级用粗线显示。靠近顶部的偏差分布表示各组的相似性 - 基本上相同的基因代表了它们各自样品中的cluster。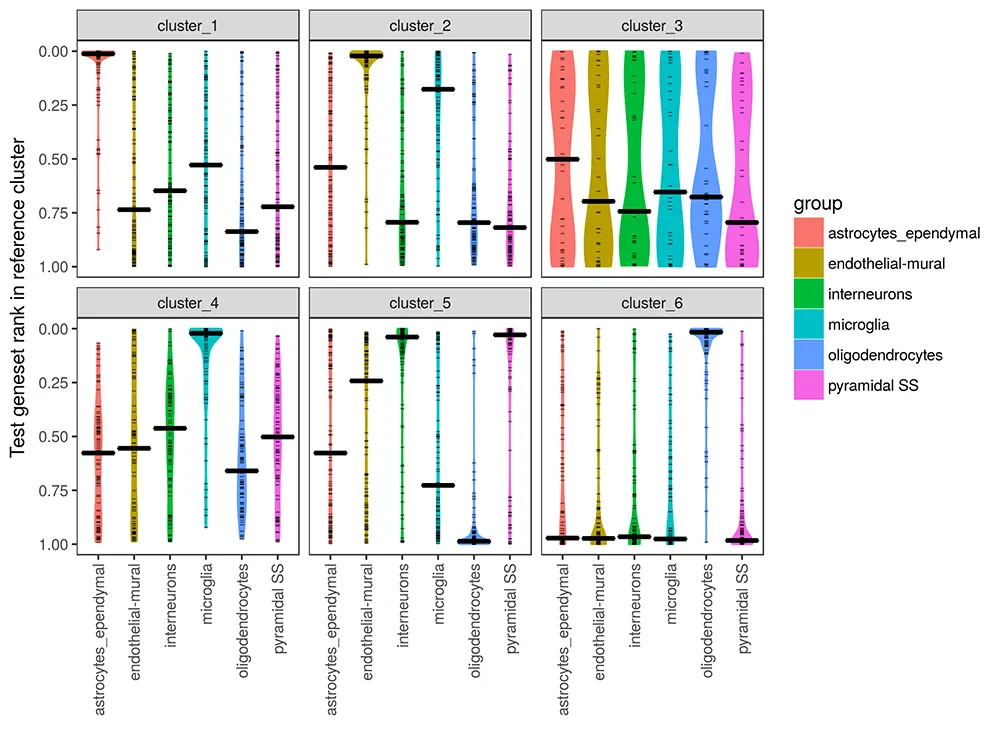
- A median gene rank of 0.5 would indicate a completely random distribution. However, much lower values are common. The reciprocal nature of the within-dataset differential expression can cause this - what’s up in one cluster is down in another.
- A small or heterogeneous cell group will not have much statistical power to select many ‘top’ genes (few tick marks) and these distributions will not be particuarly informative. If there are no ‘top’ genes it won’t be plotted at all.
- Because ‘top’ genes are compared to total reference rankings - the comparison between two datasets is not symmetrical. In ambiguous cases, it might helpful to plot the reverse comparison from reference to query. Note that these receiprocal comparisons are considered in Assigning labels to clusters. For instance - if a query cluster happens to be a mix of two reference cell groups, a reciprocal plot may make this more obvious.
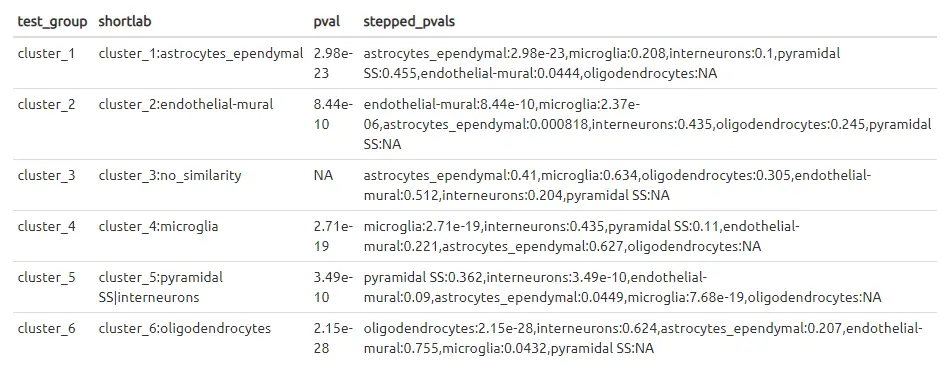
Assigning labels to clusters 为clusters分配标签
Celaref包的安装
The bioconductor landing page with information about this package is at https://bioconductor.org/packages/celaref
# To install from bioconductor via BiocManager
# Installing BiocManager if necessary:
install.packages("BiocManager")
BiocManager::install("celaref")
# Or, to use the dev version from github
devtools::install_github("MonashBioinformaticsPlatform/celaref")
Quickstart
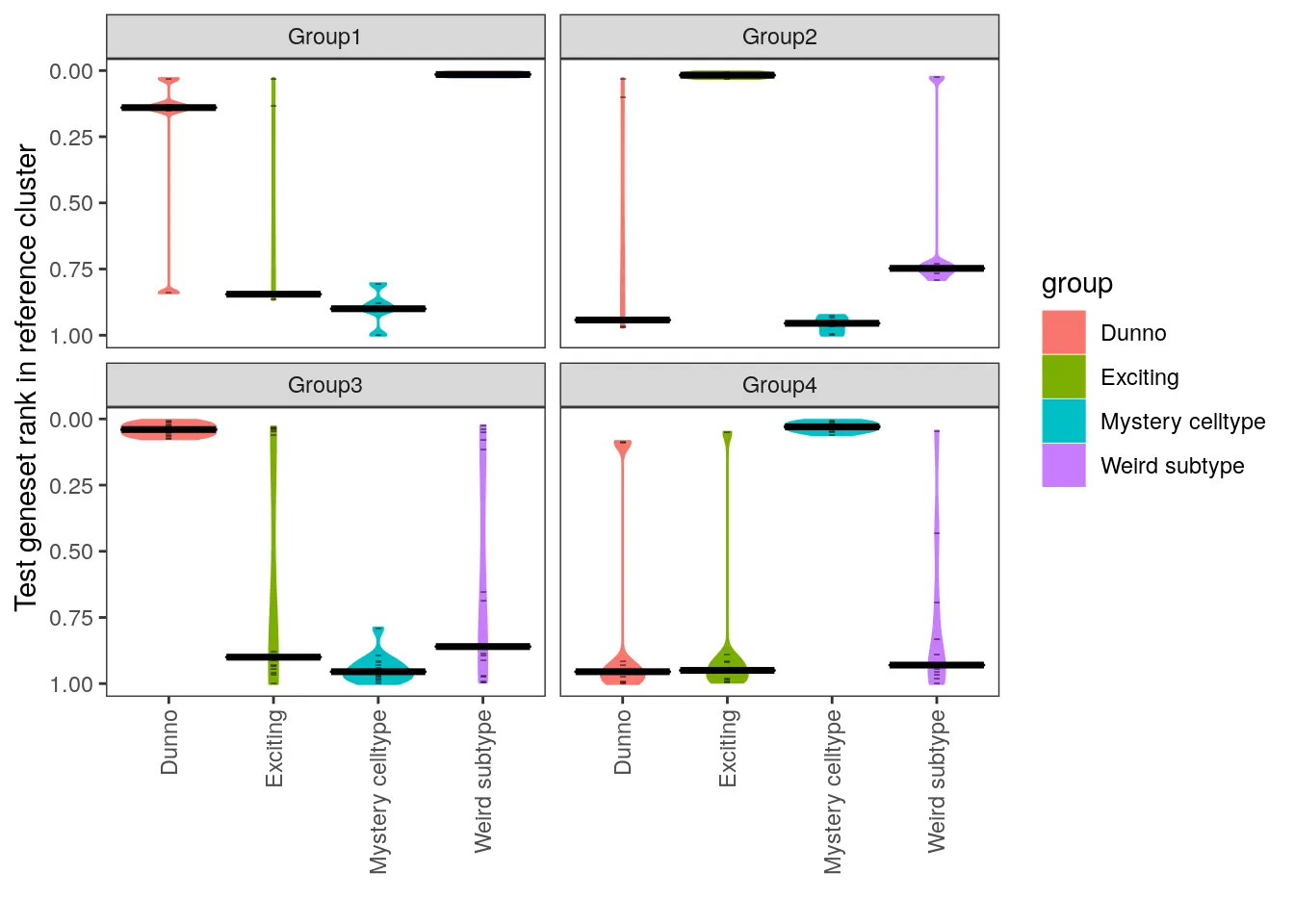
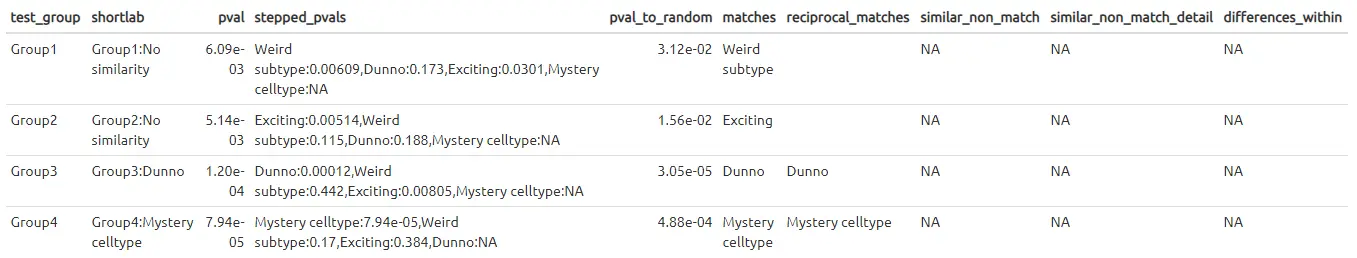
假设有一个新的scRNA-seq数据集(查询集),其细胞被分为了4个cluster组,但并不知道这些cluster分别代表哪种细胞类型。幸运的是,在相同的组织类型中,有一个其他的数据集(参考集)已经确定好了其中的细胞类型,并将其分别命名为‘Weird subtype’, ‘Exciting’, ‘Mystery cell type’和‘Dunno’。
本示例,我们将使用这个参考数据集的标签,对未知的测试数据集进行细胞类型的注释。
# 加载celaref包
library(celaref)
# Paths to data files.
# 设置查询数据集路径
counts_filepath.query <- system.file("extdata", "sim_query_counts.tab", package = "celaref")
cell_info_filepath.query <- system.file("extdata", "sim_query_cell_info.tab", package = "celaref")
# 设置参考数据集路径
counts_filepath.ref <- system.file("extdata", "sim_ref_counts.tab", package = "celaref")
cell_info_filepath.ref <- system.file("extdata", "sim_ref_cell_info.tab", package = "celaref")
# Load data
# 加载参考数据集
toy_ref_se <- load_se_from_files(counts_file=counts_filepath.ref, cell_info_file=cell_info_filepath.ref)
toy_ref_se
class: SummarizedExperiment
dim: 200 515
metadata(0):
assays(1): counts
rownames(200): Gene1 Gene2 ... Gene199 Gene200
rowData names(1): ID
colnames(515): Cell1 Cell4 ... Cell995 Cell1000
colData names(5): cell_sample Cell Batch group ExpLibSize
# 加载测试数据集
toy_query_se <- load_se_from_files(counts_file=counts_filepath.query, cell_info_file=cell_info_filepath.query)
toy_query_se
class: SummarizedExperiment
dim: 200 485
metadata(0):
assays(1): counts
rownames(200): Gene1 Gene2 ... Gene199 Gene200
rowData names(1): ID
colnames(485): Cell2 Cell3 ... Cell998 Cell999
colData names(5): cell_sample Cell Batch group ExpLibSize
# Filter data
# 使用trim_small_groups_and_low_expression_genes函数进行数据过滤
toy_ref_se <- trim_small_groups_and_low_expression_genes(toy_ref_se)
toy_query_se <- trim_small_groups_and_low_expression_genes(toy_query_se)
# Setup within-experiment differential expression
# 使用contrast_each_group_to_the_rest函数进行差异表达分析
de_table.toy_ref <- contrast_each_group_to_the_rest(toy_ref_se, dataset_name="ref")
head(de_table.toy_ref)[,1:5]
de_table.toy_query <- contrast_each_group_to_the_rest(toy_query_se, dataset_name="query")
head(de_table.toy_query)[,1:5]
# Plot
# 使用make_ranking_violin_plot函数可视化分析结果
make_ranking_violin_plot(de_table.test=de_table.toy_query, de_table.ref=de_table.toy_ref)
#> Warning: `fun.y` is deprecated. Use `fun` instead.
#> Warning: `fun.ymin` is deprecated. Use `fun.min` instead.
#> Warning: `fun.ymax` is deprecated. Use `fun.max` instead.
# And get group labels
# 使用make_ref_similarity_names函数查看注释的结果
make_ref_similarity_names(de_table.toy_query, de_table.toy_ref)
参考来源:http://www.bioconductor.org/packages/release/bioc/vignettes/celaref/inst/doc/celaref_doco.html

若有收获,就点个赞吧
0 人点赞
