第八节:拟时序细胞轨迹推断
在本节教程中,我们将学习如何通过拟时序分析推断细胞分化轨迹。slingshot包可以对单细胞RNA-seq数据进行细胞分化谱系构建和伪时间推断,它利用细胞聚类簇和空间降维信息,以无监督或半监督的方式学习细胞聚类群之间的关系,揭示细胞聚类簇之间的全局结构,并将该结构转换为由一维变量表示的平滑谱系,称之为“伪时间”。
运行slingshot的至少需要两个输入文件:即细胞在降维空间中的坐标矩阵和细胞聚类群的标签向量。通过这两个输入文件,我们可以:
- 使用
getLineages函数在细胞聚类群上构建最小生成树(MST),确定细胞全局的谱系结构; - 利用
getCurves函数拟合主曲线来构造平滑谱系,并推断伪时间变量; - 使用内置的可视化工具评估不同步骤的分析结果。
安装并加载所需的R包
BiocManager::install("slingshot")library(slingshot)library(SingleCellExperiment)
构建测试数据集
这里,我们模拟了两个不同形式的数据集进行谱系推断分析。
接下来,我们构建第一个测试数据集,用于表示单个谱系,其中三分之一的基因与过渡相关。我们将该数据集包含在一个SingleCellExperiment对象中,并将其用于演示相应的分析流程。
# generate synthetic count data representing a single lineage
# 构建单细胞表达矩阵
means <- rbind(
# non-DE genes
matrix(rep(rep(c(0.1,0.5,1,2,3), each = 300),100),
ncol = 300, byrow = TRUE),
# early deactivation
matrix(rep(exp(atan( ((300:1)-200)/50 )),50), ncol = 300, byrow = TRUE),
# late deactivation
matrix(rep(exp(atan( ((300:1)-100)/50 )),50), ncol = 300, byrow = TRUE),
# early activation
matrix(rep(exp(atan( ((1:300)-100)/50 )),50), ncol = 300, byrow = TRUE),
# late activation
matrix(rep(exp(atan( ((1:300)-200)/50 )),50), ncol = 300, byrow = TRUE),
# transient
matrix(rep(exp(atan( c((1:100)/33, rep(3,100), (100:1)/33) )),50),
ncol = 300, byrow = TRUE)
)
counts <- apply(means,2,function(cell_means){
total <- rnbinom(1, mu = 7500, size = 4)
rmultinom(1, total, cell_means)
})
rownames(counts) <- paste0('G',1:750)
colnames(counts) <- paste0('c',1:300)
counts[1:5,1:5]
# c1 c2 c3 c4 c5
#G1 0 2 0 0 0
#G2 5 2 2 2 9
#G3 5 9 3 5 8
#G4 6 7 5 12 23
#G5 14 21 12 17 20
dim(counts)
#[1] 750 300
# 将表达矩阵转换为SingleCellExperiment对象
sim <- SingleCellExperiment(assays = List(counts = counts))
sim
#class: SingleCellExperiment
#dim: 750 300
#metadata(0):
#assays(1): counts
#rownames(750): G1 G2 ... G749 G750
#rowData names(0):
#colnames(300): c1 c2 ... c299 c300
#colData names(0):
#reducedDimNames(0):
#spikeNames(0):
接下来,我们构建第二个示例数据集,该数据集由降维空间坐标矩阵(如通过PCA, ICA和diffusion map等获得)和细胞聚类群标签(如K-means聚类等生成)组成。该数据集表示一个分叉的轨迹,可以用来演示slingshot包的其他功能。
# 加载slingshot包内置数据
data("slingshotExample")
rd <- slingshotExample$rd
cl <- slingshotExample$cl
dim(rd) # data representing cells in a reduced dimensional space
## [1] 140 2
length(cl) # vector of cluster labels
## [1] 140
head(rd)
# dim-1 dim-2
#cell-1 -4.437117 0.3690586
#cell-2 -3.837149 -0.1009774
#cell-3 -2.730936 -0.9770827
#cell-4 -4.219531 -0.7100939
#cell-5 -3.717672 -0.6452345
#cell-6 -3.053360 -0.2838916
head(cl)
#cell-1 cell-2 cell-3 cell-4 cell-5 cell-6
# 1 1 1 1 1 1
上游分析
基因过滤
要开始对单个谱系数据集进行分析,我们需要降低数据的维数,并过滤掉无信息的基因。这将大大提高下游分析的速度,同时将信息损失降至最低。
对于基因过滤步骤,我们保留了至少在10个细胞中表达,且基因表达count数至少大于3的基因。
# filter genes down to potential cell-type markers
# at least M (15) reads in at least N (15) cells
geneFilter <- apply(assays(sim)$counts,1,function(x){
sum(x >= 3) >= 10
})
sim <- sim[geneFilter, ]
数据标准化
由于我们使用的是模拟数据,因此无需担心批处理效应或其他潜在的混杂因素。在这里,我们将使用分位数归一化的方法对表达数据进行归一化处理,它可以使每个细胞具有相同的表达值分布。
FQnorm <- function(counts){
rk <- apply(counts,2,rank,ties.method='min')
counts.sort <- apply(counts,2,sort)
refdist <- apply(counts.sort,1,median)
norm <- apply(rk,2,function(r){ refdist[r] })
rownames(norm) <- rownames(counts)
return(norm)
}
assays(sim)$norm <- FQnorm(assays(sim)$counts)
数据降维
slingshot基本假设是,转录相似的细胞在某些降维空间中会彼此靠近。由于我们在构造谱系和测量伪时间时使用欧几里得距离,因此对数据进行降维处理很重要。
在这里,我们将演示两种降维方法:主成分分析(PCA)和均匀流形逼近和投影(UMAP,通过uwot包调用)。
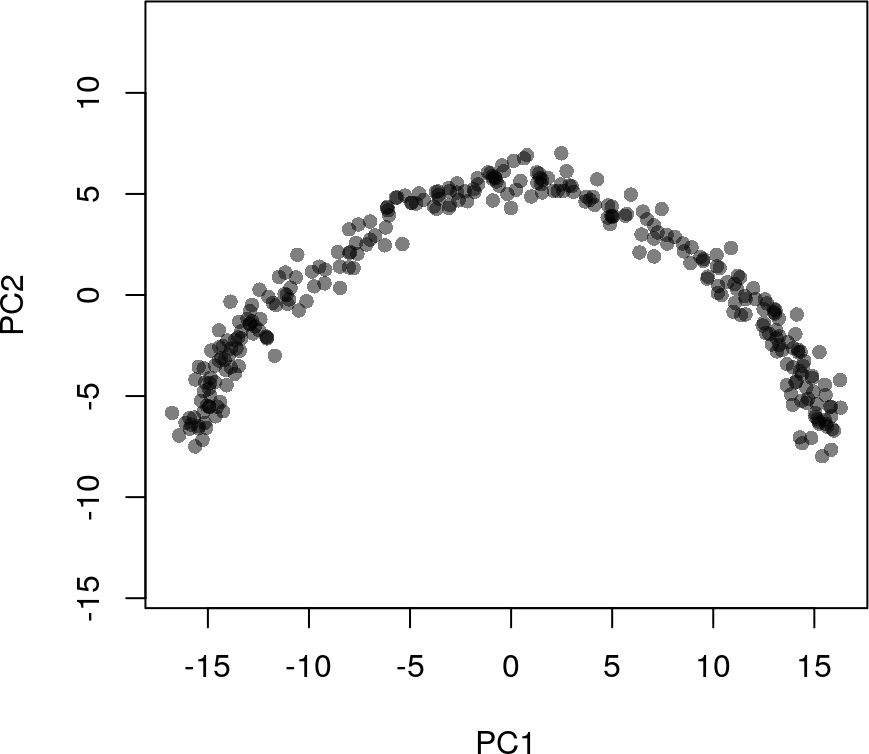
# 使用prcomp函数进行PCA降维
pca <- prcomp(t(log1p(assays(sim)$norm)), scale. = FALSE)
rd1 <- pca$x[,1:2]
head(rd1)
# PC1 PC2
#c1 -15.55731 -6.218455
#c2 -14.48979 -7.060021
#c3 -16.10326 -7.630399
#c4 -15.59147 -7.443861
#c5 -15.86607 -6.180724
#c6 -15.78163 -6.733366
plot(rd1, col = rgb(0,0,0,.5), pch=16, asp = 1)
library(uwot)
## Loading required package: Matrix
##
## Attaching package: 'Matrix'
## The following object is masked from 'package:S4Vectors':
##
## expand
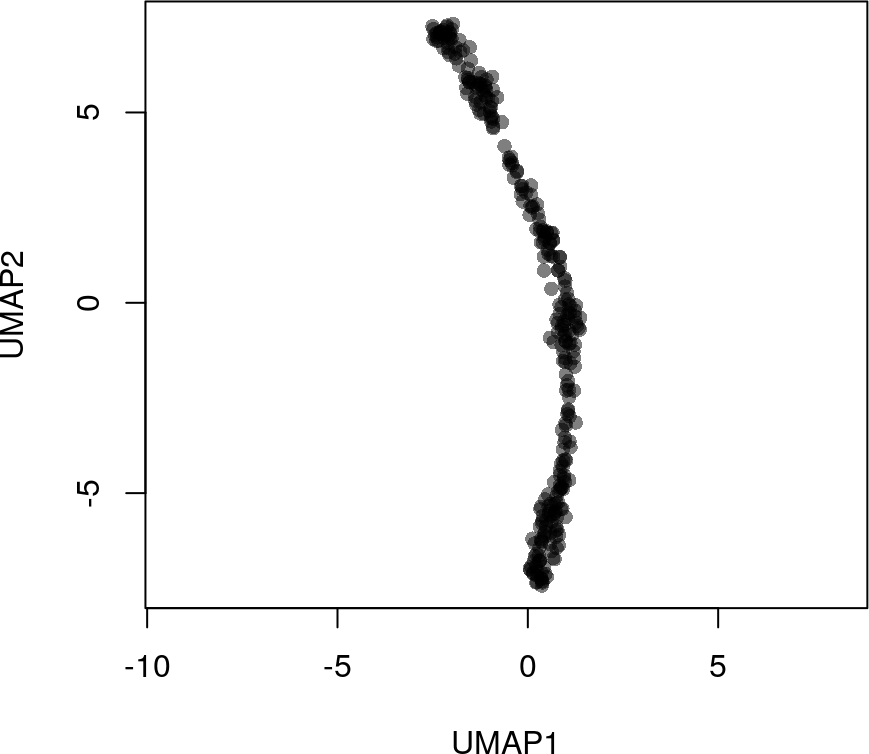
# 进行UMAP降维
rd2 <- umap(t(log1p(assays(sim)$norm)))
colnames(rd2) <- c('UMAP1', 'UMAP2')
head(rd2)
# UMAP1 UMAP2
#[1,] 6.357223 -4.358757
#[2,] 6.442034 -4.216842
#[3,] 6.478745 -4.421883
#[4,] 6.712623 -4.487934
#[5,] 6.497382 -4.276127
#[6,] 6.302136 -4.531643
plot(rd2, col = rgb(0,0,0,.5), pch=16, asp = 1)
接下来,我们可以将降维的结果添加到SingleCellExperiment对象中。
reducedDims(sim) <- SimpleList(PCA = rd1, UMAP = rd2)
sim
#class: SingleCellExperiment
#dim: 734 300
#metadata(0):
#assays(2): counts norm
#rownames(734): G2 G3 ... G749 G750
#rowData names(0):
#colnames(300): c1 c2 ... c299 c300
#colData names(0):
#reducedDimNames(2): PCA UMAP
#spikeNames(0):
细胞聚类
slingshot的最终输入是细胞聚类簇的标签向量。如果未提供此参数,slingshot则将数据视为单个细胞簇并拟合标准主曲线。但是,我们建议即使在仅期望单一谱系的数据集中也应该对细胞进行聚类分群,因为它可以潜在地发现新的分支事件。
在此步骤中得到的聚类群集将用于确定潜在分化谱系的全局结构。这与聚类单细胞数据的典型目标不同,后者的主要目的是识别数据集中存在的所有生物学相关的细胞类型。例如,在确定全局谱系结构时,无需区分未成熟神经元和成熟神经元,因为两种细胞类型可能会沿着谱系的同一段落入。
在这里,我们采用了两种聚类方法:高斯混合建模和 k-means聚类,它们类似地假设低维空间中的欧几里得距离反映了细胞之间的生物学差。前者可以使用mclust包进行实现(Scrucca et al,2016年),它可以提供一种基于贝叶斯信息准则(BIC)的方法自动确定细胞聚类的簇数。
library(mclust, quietly = TRUE)
## Package 'mclust' version 5.4.6
## Type 'citation("mclust")' for citing this R package in publications.
##
## Attaching package: 'mclust'
## The following object is masked from 'package:mgcv':
##
## mvn
# 使用Mclust函数进行细胞聚类
cl1 <- Mclust(rd1)$classification
head(cl1)
#c1 c2 c3 c4 c5 c6
# 1 1 1 1 1 1
colData(sim)$GMM <- cl1
sim
#class: SingleCellExperiment
#dim: 734 300
#metadata(0):
#assays(2): counts norm
#rownames(734): G2 G3 ... G749 G750
#rowData names(0):
#colnames(300): c1 c2 ... c299 c300
#colData names(1): GMM
#reducedDimNames(2): PCA UMAP
#spikeNames(0):
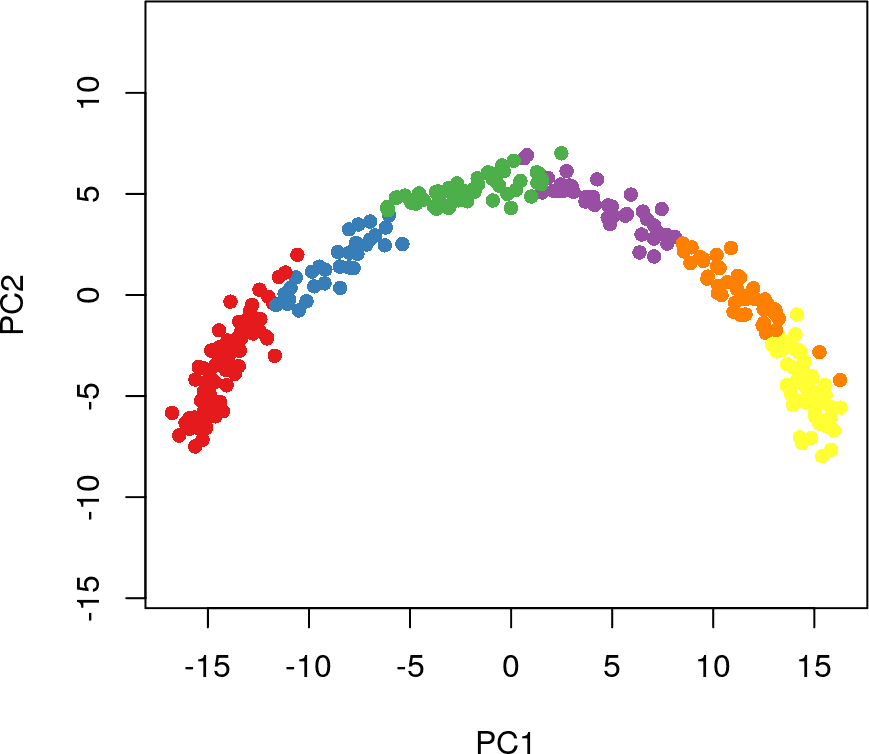
# 可视化聚类分群的结果
library(RColorBrewer)
plot(rd1, col = brewer.pal(9,"Set1")[cl1], pch=16, asp = 1)
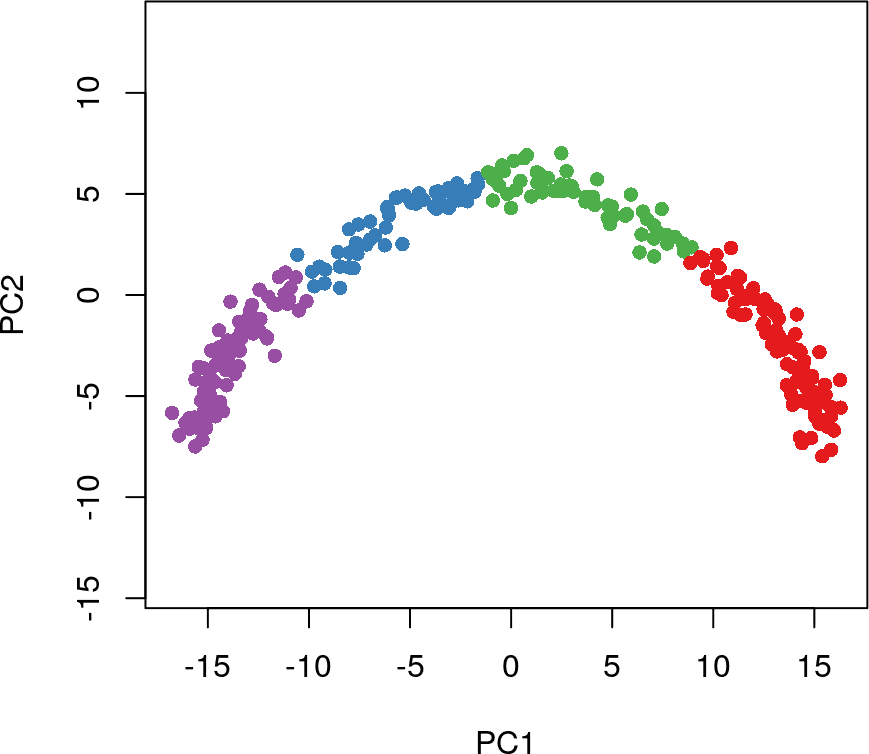
k-means聚类没有类似的功能,因此我们需要人为的指定聚类的细胞簇数。如果细胞簇数选择的太低,我们可能会错过一个真正的分支事件;如果它太高或有很多小簇,我们可能会开始看到虚假的分支事件。这里,我们选择聚类簇数为4进行k-means聚类分析。
# 使用kmeans函数进行细胞聚类
cl2 <- kmeans(rd1, centers = 4)$cluster
head(cl2)
#c1 c2 c3 c4 c5 c6
# 2 2 2 2 2 2
colData(sim)$kmeans <- cl2
# 可视化聚类分群的结果
plot(rd1, col = brewer.pal(9,"Set1")[cl2], pch=16, asp = 1)
使用slingshot构建分化谱系并进行拟时推断
现在,我们已经得到了slingshot分析所要用的两个文件:细胞降维的空间坐标矩阵和聚类分群的标签向量。接下来,我们就可以运行slinghsot进行伪时间推断分析。这是一个两步的过程,包括用基于细胞群集的最小生成树(MST)识别全局谱系结构,以及拟合主要曲线来描述每个谱系。
这两个步骤可以使用getLineages和getCurves函数分开运行,也可以使用slingshot封装函数一起运行(推荐)。这里,我们将使用封装函数对单轨迹数据集进行谱系分析,稍后将在分叉数据集上演示分步函数的用法。
为了使用PCA降维产生的空间坐标信息和高斯混合建模识别的细胞聚类簇标签来运行slingshot,我们将执行以下操作:
sim <- slingshot(sim, clusterLabels = 'GMM', reducedDim = 'PCA')
## Using full covariance matrix
sim
class: SingleCellExperiment
dim: 734 300
metadata(0):
assays(2): counts norm
rownames(734): G2 G3 ... G749 G750
rowData names(0):
colnames(300): c1 c2 ... c299 c300
colData names(3): GMM kmeans slingPseudotime_1
reducedDimNames(2): PCA UMAP
spikeNames(0):
如上所述,如果未提供聚类分群信息,slingshot则假定所有细胞都是同一个聚类群,并且将构建一条拟合曲线。如果未提供降维空间坐标信息,slingshot将使用由返回的列表的第一个元素reducedDims中的信息。
输出的结果是一个包含了slingshot结果的SingleCellExperiment对象。大多数的输出以列表的形式添加到元数据中,并可以通过metadata(sce)$slingshot来进行访问。此外,所有推断的伪时间变量(每个谱系一个)都添加到colData中。如果要提取slingshot单个对象中的所有结果,我们可以使用SlingshotDataSet函数。这对于可视化很有用,因为SlingshotDataSet函数中包含了几种对象绘制方法 。下面,我们对单轨迹数据谱系推断的结果进行可视化处理,并用伪时间对细胞进行着色。
summary(sim$slingPseudotime_1)
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.000 8.633 21.118 21.415 34.367 43.186
colors <- colorRampPalette(brewer.pal(11,'Spectral')[-6])(100)
plotcol <- colors[cut(sim$slingPseudotime_1, breaks=100)]
plot(reducedDims(sim)$PCA, col = plotcol, pch=16, asp = 1)
lines(SlingshotDataSet(sim), lwd=2, col='black')
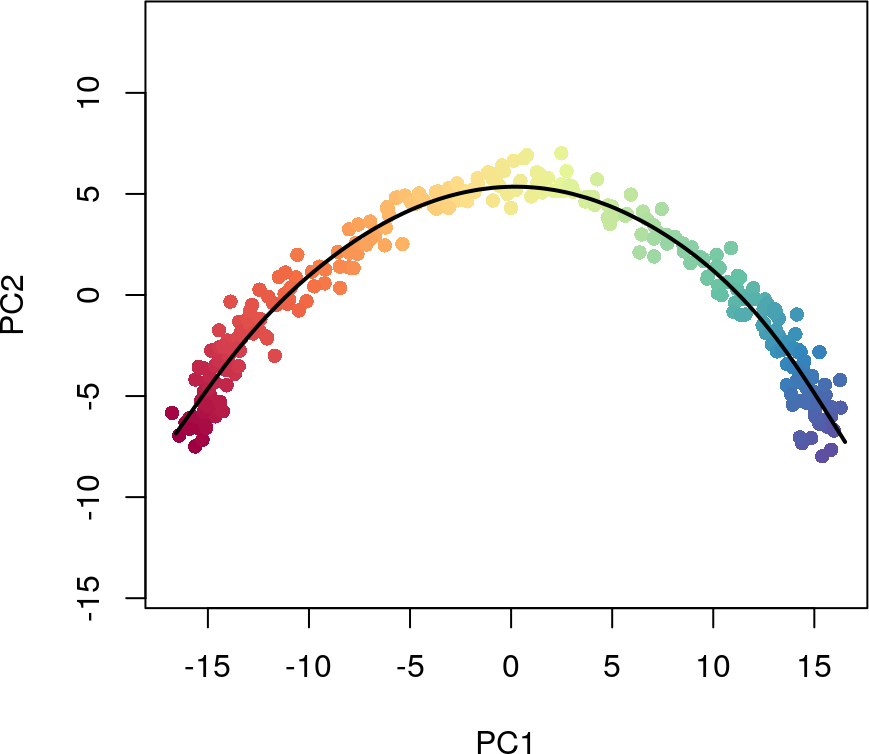
通过使用type参数,还可以看到基于细胞聚类群的最小生成树是如何推断出谱系结构的。
plot(reducedDims(sim)$PCA, col = brewer.pal(9,'Set1')[sim$GMM], pch=16, asp = 1)
lines(SlingshotDataSet(sim), lwd=2, type = 'lineages', col = 'black')
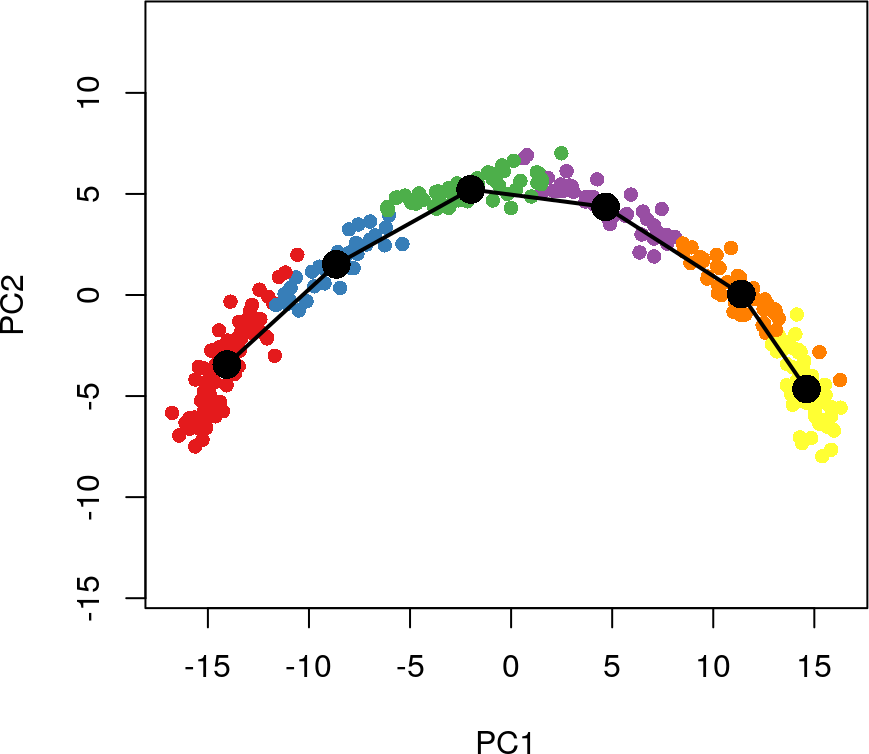
下游分析
鉴定随伪时间表达变化的基因
运行完slingshot推断出细胞分化谱系后,我们可以进一步鉴定出随伪时间或发育过程中表达变化的基因。我们将使用tradeSeq软件包演示这种类型的分析(Van den Berge等,2020)。
对于每个基因,我们将使用负二项式噪声分布来拟合通用加性模型(GAM),以对基因表达与伪时间之间的(可能是非线性的)关系进行建模。然后,我们使用 associationTest函数来鉴定与伪时间存在显著关联的基因。
BiocManager::install("tradeSeq")
library(tradeSeq)
# fit negative binomial GAM
sim <- fitGAM(sim)
# test for dynamic expression
ATres <- associationTest(sim)
然后,我们可以根据p值挑选出变化最显著的基因,并通过热图可视化其在整个伪时间或发育过程中的表达情况。这里,我们使用top250基因进行展示。
topgenes <- rownames(ATres[order(ATres$pvalue), ])[1:250]
pst.ord <- order(sim$slingPseudotime_1, na.last = NA)
heatdata <- assays(sim)$counts[topgenes, pst.ord]
heatclus <- sim$GMM[pst.ord]
heatmap(log1p(heatdata), Colv = NA,
ColSideColors = brewer.pal(9,"Set1")[heatclus])
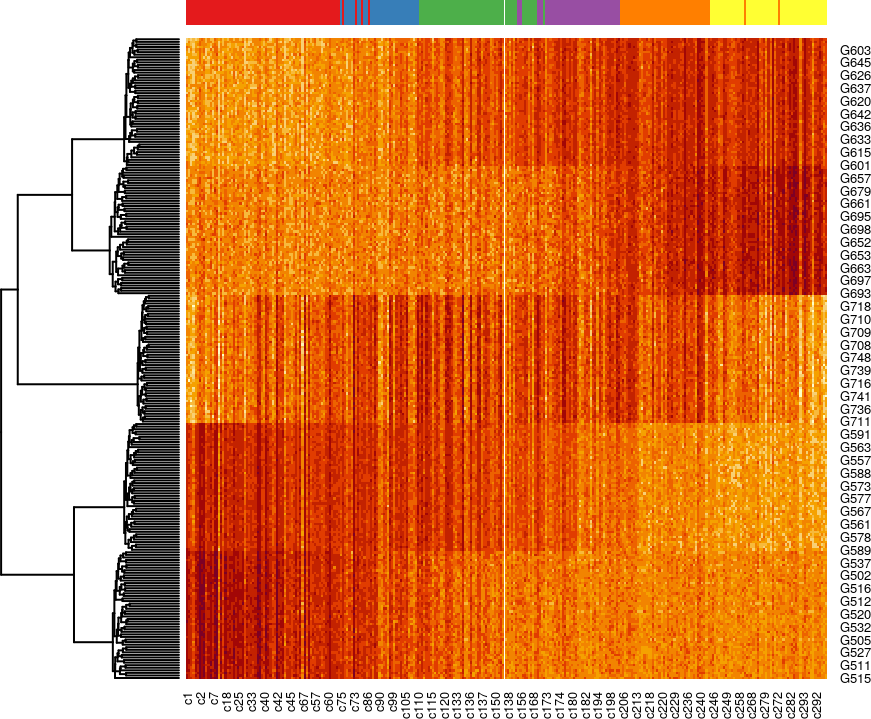
分步运行slingshot
接下来,我们将重点介绍slingshot包的一些其他功能。我们使用该包内置的slingshotExample数据集进行说明。此数据集中包含了细胞降维空间的低维表示,以及k-means聚类后的细胞簇标签。
确定细胞分化谱系的全局结构
首先,我们getLineages函数构建细胞分化谱系的全局结构。该函数使用n × p的细胞降维空间矩阵和聚类分群的标签向量n作为输入,通过最小生成树(MST)算法映射相邻细胞群集之间的连接,并进一步通过这些连接关系鉴定出细胞分化谱系的路径。该函数的输出结果是一个 SlingshotDataSet对象,其中包含了输入文件,以及推断出的MST(由邻接矩阵表示)和分化谱系(细胞聚类簇的有序向量)。
通过指定已知的起始簇和终点簇,我们还可以以完全无监督的方式或以半监督的方式执行此分析。如果我们不指定起点,则slingshot根据简约性选择一个起点,以使分割前谱系之间共享的细胞簇数最大化。如果没有分割或多个簇产生相同的简约分数,则可以任意选择起始簇。
对于我们的模拟数据,这里选择以“Cluster1”作为起始簇。但是,我们通常建议根据先验知识(样本采集的时间或已知的基因标记)确定初始簇的类别。
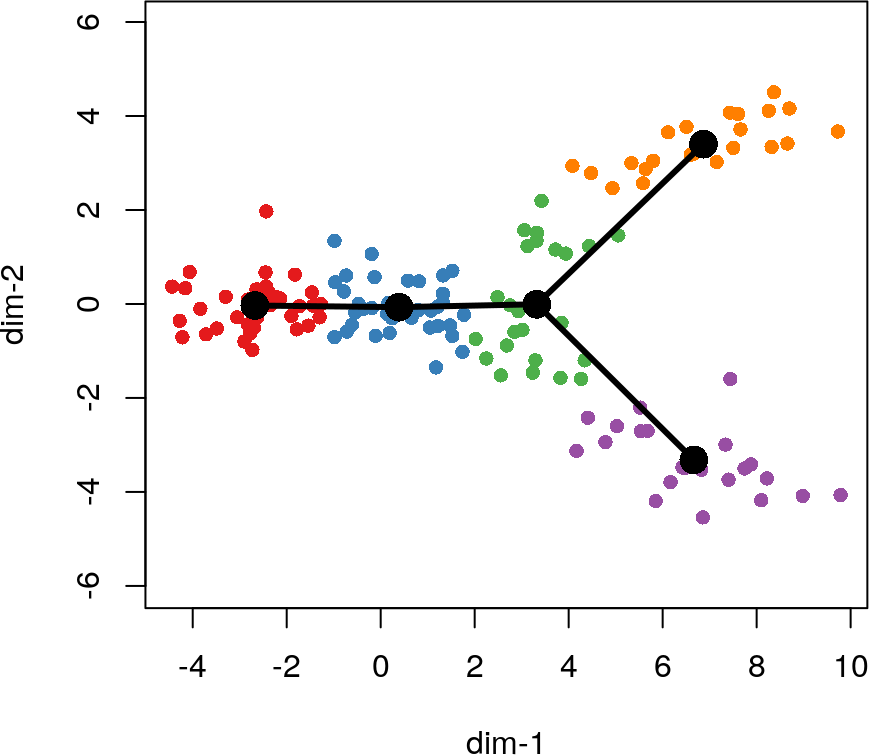
lin1 <- getLineages(rd, cl, start.clus = '1')
## Using full covariance matrix
lin1
## class: SlingshotDataSet
##
## Samples Dimensions
## 140 2
##
## lineages: 2
## Lineage1: 1 2 3 5
## Lineage2: 1 2 3 4
##
## curves: 0
plot(rd, col = brewer.pal(9,"Set1")[cl], asp = 1, pch = 16)
lines(lin1, lwd = 3, col = 'black')
在此步骤中,我们还可以指定已知的终点簇。当构造MST时,被指定为终端状态的细胞簇将被约束为仅具有一个连接(即,它们必须是叶节点)。该约束可能会影响树的其他部分的绘制方式,如以下示例中所示,在该示例中,我们将Cluster3指定为终点簇。
lin2 <- getLineages(rd, cl, start.clus= '1', end.clus = '3')
## Using full covariance matrix
plot(rd, col = brewer.pal(9,"Set1")[cl], asp = 1, pch = 16)
lines(lin2, lwd = 3, col = 'black', show.constraints = TRUE)
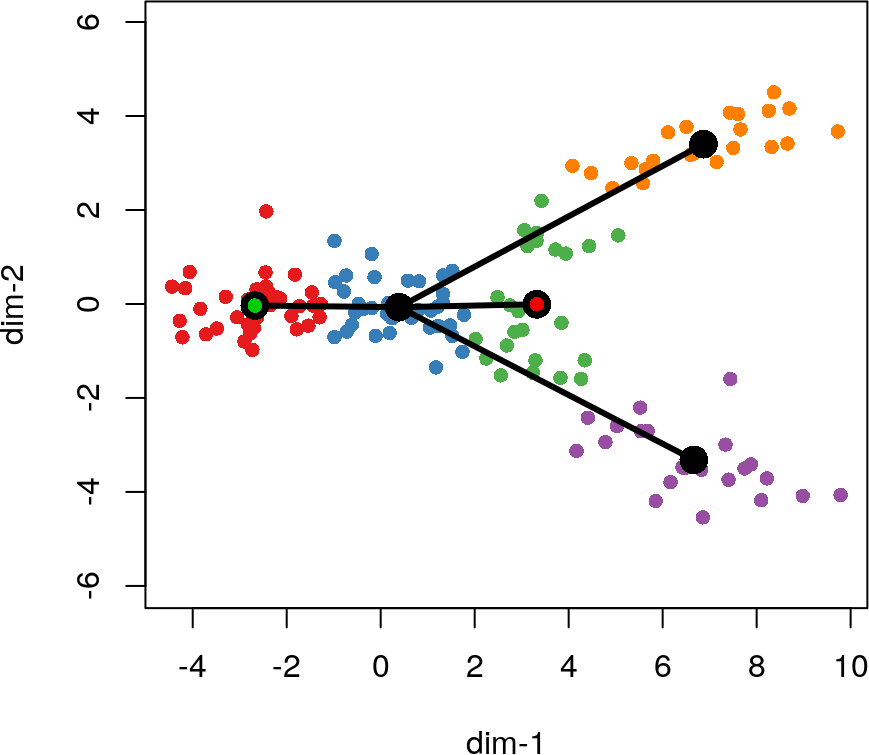
这种类型的监督对于确保结果与先前的生物学知识一致可能是有用的。具体地,它可以防止将已知的终末细胞命运归为暂时状态。
我们还可以传递一些其他参数,以实现对getLineages函数更好的控制:
dist.funis a function for computing distances between clusters. The default is squared distance between cluster centers normalized by their joint covariance matrix.omegais a granularity parameter, allowing the user to set an upper limit on connection distances. This can be useful for identifying outlier clusters which are not a part of any lineage or for separating distinct trajectories. This is typically a numeric argument, but a value of TRUE will indicate that a heuristic of 3 * (median edge length of unsupervised MST) should be used.
构建平滑曲线确定细胞分化顺序
为了构建平滑的谱系曲线,我们使用getCurves函数对getLineages的结果进行处理。该函数遵循类似于(Hastie and Stuetzle 1989)中提出的主要曲线的迭代过程。如果只有一个分化谱系,则生成的曲线只是经过数据中心的主曲线,并进行了一次调整:初始曲线是通过聚类中心之间的线性连接而不是数据的第一个主成分而构建的。这种调整增加了稳定性,并通常加快了算法的收敛速度。
当有两个或多个分化谱系时,我们向该算法添加一个额外的步骤:对共享细胞附近的曲线求平均。两种谱系应该在尚未分化的细胞上都很好地吻合,因此在每次迭代中,我们均对这些细胞附近的曲线进行平均。这增加了算法的稳定性,并产生了平滑的分支谱系。
crv1 <- getCurves(lin1)
crv1
## class: SlingshotDataSet
##
## Samples Dimensions
## 140 2
##
## lineages: 2
## Lineage1: 1 2 3 5
## Lineage2: 1 2 3 4
##
## curves: 2
## Curve1: Length: 15.045 Samples: 100.6
## Curve2: Length: 15.126 Samples: 103.5
plot(rd, col = brewer.pal(9,"Set1")[cl], asp = 1, pch = 16)
lines(crv1, lwd = 3, col = 'black')
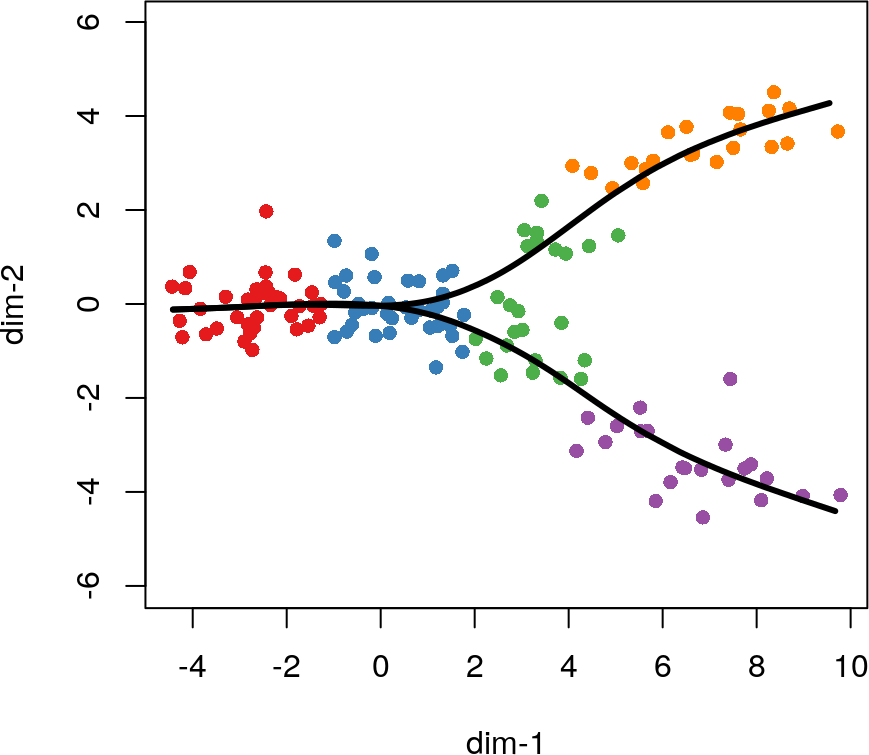
getCurves函数的结果也是一个slingshotDataSet对象,其中包含了分化谱系的主曲线和有关其拟合方式的其他信息。我们可以使用pseudotime函数提取不同分化谱系中细胞的伪时间值,其中的NA表示未分配给特定谱系的细胞的值。使用curveWeights函数可以提取相似的权重矩阵,将每个细胞分配给一个或多个谱系。
可以使用curves函数访问曲线对象,该函数将返回principal_curve对象的列表。这些对象中包含以下信息:
s: the matrix of points that make up the curve. These correspond to the orthogonal projections of the data points.ord: indices which can be used to put the cells along a curve in order based their projections.
lambda: arclengths along the curve from the beginning to each cell’s projection. A matrix of these values is returned by the slingPseudotime function.dist_ind: the squared distances between data points and their projections onto the curve.dist: the sum of the squared projection distances.w: the vector of weights along this lineage. Cells that were unambiguously assigned to this lineage will have a weight of 1, while cells assigned to other lineages will have a weight of 0. It is possible for cells to have weights of 1 (or very close to 1) for multiple lineages, if they are positioned before a branching event. A matrix of these values is returned by the slingCurveWeights function.
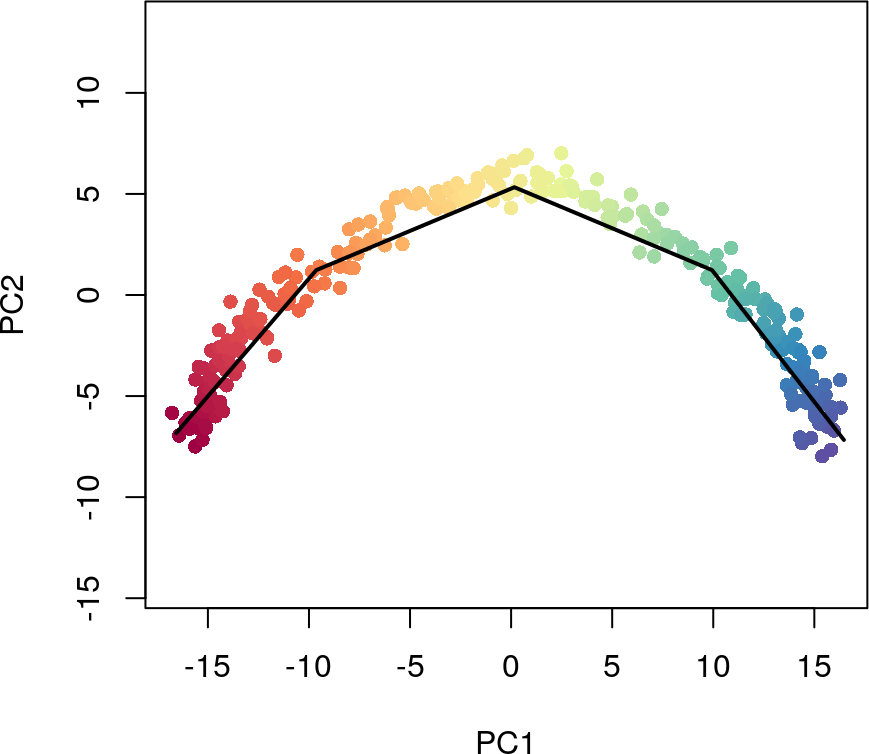
在大型数据集上运行slingshot
对于大型数据集,我们建议在运行slingshot函数时使用approx_points参数。通过设置approx_points,用户可以指定曲线的分辨率(唯一点的数量),这可以大大缩短计算时间,并且精度损失最小。我们建议该值设置为100-200。
sim5 <- slingshot(sim, clusterLabels = 'GMM', reducedDim = 'PCA',
approx_points = 5)
## Using full covariance matrix
colors <- colorRampPalette(brewer.pal(11,'Spectral')[-6])(100)
plotcol <- colors[cut(sim5$slingPseudotime_1, breaks=100)]
plot(reducedDims(sim5)$PCA, col = plotcol, pch=16, asp = 1)
lines(SlingshotDataSet(sim5), lwd=2, col='black')
sessionInfo()
## R version 4.0.3 (2020-10-10)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 18.04.5 LTS
##
## Matrix products: default
## BLAS: /home/biocbuild/bbs-3.12-bioc/R/lib/libRblas.so
## LAPACK: /home/biocbuild/bbs-3.12-bioc/R/lib/libRlapack.so
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=C
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] parallel stats4 stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] mclust_5.4.6 uwot_0.1.8
## [3] Matrix_1.2-18 SingleCellExperiment_1.12.0
## [5] SummarizedExperiment_1.20.0 Biobase_2.50.0
## [7] GenomicRanges_1.42.0 GenomeInfoDb_1.26.0
## [9] IRanges_2.24.0 S4Vectors_0.28.0
## [11] BiocGenerics_0.36.0 MatrixGenerics_1.2.0
## [13] matrixStats_0.57.0 mgcv_1.8-33
## [15] nlme_3.1-150 RColorBrewer_1.1-2
## [17] slingshot_1.8.0 princurve_2.1.5
## [19] BiocStyle_2.18.0
##
## loaded via a namespace (and not attached):
## [1] Rcpp_1.0.5 RSpectra_0.16-0 compiler_4.0.3
## [4] BiocManager_1.30.10 XVector_0.30.0 bitops_1.0-6
## [7] tools_4.0.3 zlibbioc_1.36.0 digest_0.6.27
## [10] evaluate_0.14 lattice_0.20-41 pkgconfig_2.0.3
## [13] rlang_0.4.8 igraph_1.2.6 DelayedArray_0.16.0
## [16] magick_2.5.0 yaml_2.2.1 xfun_0.18
## [19] GenomeInfoDbData_1.2.4 stringr_1.4.0 knitr_1.30
## [22] grid_4.0.3 rmarkdown_2.5 bookdown_0.21
## [25] magrittr_1.5 codetools_0.2-16 splines_4.0.3
## [28] htmltools_0.5.0 ape_5.4-1 stringi_1.5.3
## [31] RCurl_1.98-1.2 FNN_1.1.3
参考来源:https://bioconductor.org/packages/release/bioc/vignettes/slingshot/inst/doc/vignette.html

若有收获,就点个赞吧
0 人点赞
