- 简介
- 分析流程
- Step 0. Download data
- Step 1. Create snap object
- Step 2. Select barcode
- Step 3. Add cell-by-bin matrix
- Step 4. Combine snap objects
- Step 5. Binarize matrix
- Step 6. Filter bins
- Step 7. Reduce dimensionality
- Step 8. Determine significant components
- Step 9. Remove batch effect
- Step 10. Graph-based cluster
- Step 11. Visualization
简介
在本教程中,我们将对来自10X和snATAC-seq技术产生的成年小鼠大脑的单细胞ATAC-seq测序数据进行整合分析。该示例的所有数据可以从以下链接进行下载:http://renlab.sdsc.edu/r3fang/share/github/Mouse_Brain_10X_snATAC/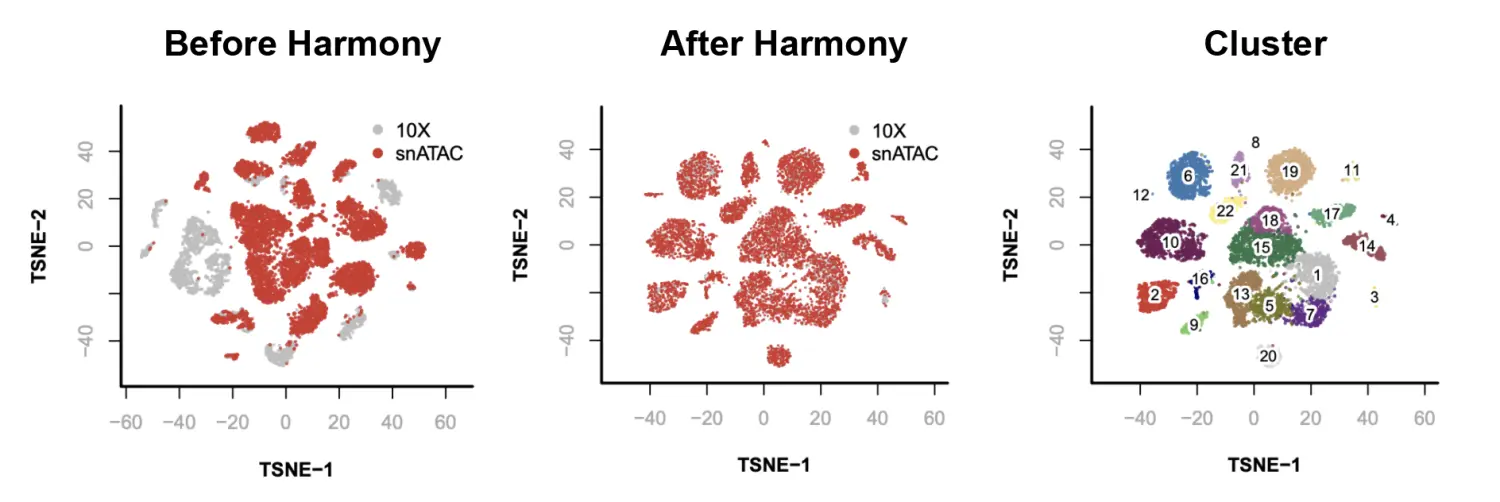
分析流程
- Step 0. Download data
- Step 1. Create snap object
- Step 2. Select barcode
- Step 3. Add cell-by-bin matrix
- Step 4. Combine snap objects
- Step 5. Filter bins
- Step 6. Dimensionality reduction
- Step 7. Determine significant components
- Step 8. Remove batch effect
- Step 9. Graph-based cluster
- Step 10. Visualization
Step 0. Download data
# 下载所需的数据集$ wget http://renlab.sdsc.edu/r3fang/share/github/Mouse_Brain_10X_snATAC/CEMBA180305_2B.snap$ wget http://renlab.sdsc.edu/r3fang/share/github/Mouse_Brain_10X_snATAC/CEMBA180305_2B.barcode.txt$ wget http://renlab.sdsc.edu/r3fang/share/github/Mouse_Brain_10X_snATAC/atac_v1_adult_brain_fresh_5k.snap$ wget http://renlab.sdsc.edu/r3fang/share/github/Mouse_Brain_10X_snATAC/atac_v1_adult_brain_fresh_5k.barcode.txt
Step 1. Create snap object
首先,我们将所用的两个数据集读取到snap对象列表中。
# 加载SnapATAC包> library(SnapATAC);> file.list = c("CEMBA180305_2B.snap", "atac_v1_adult_brain_fresh_5k.snap");> sample.list = c("snATAC", "10X");# 读取snap文件> x.sp.ls = lapply(seq(file.list), function(i){x.sp = createSnap(file=file.list[i], sample=sample.list[i]);x.sp})> names(x.sp.ls) = sample.list;# 查看snap文件信息> x.sp.ls## $snATAC## number of barcodes: 15136## number of bins: 0## number of genes: 0## number of peaks: 0## number of motifs: 0#### $`10X`## number of barcodes: 20000## number of bins: 0## number of genes: 0## number of peaks: 0## number of motifs: 0
Step 2. Select barcode
接下来,我们将读取这两个数据集的barcode信息,并选择高质量的barcodes。
> barcode.file.list = c("CEMBA180305_2B.barcode.txt", "atac_v1_adult_brain_fresh_5k.barcode.txt");# 读取barcode信息> barcode.list = lapply(barcode.file.list, function(file){read.table(file)[,1];})> x.sp.list = lapply(seq(x.sp.ls), function(i){x.sp = x.sp.ls[[i]];x.sp = x.sp[x.sp@barcode %in% barcode.list[[i]],];})> names(x.sp.list) = sample.list;> x.sp.list## $snATAC## number of barcodes: 9646## number of bins: 0## number of genes: 0## number of peaks: 0## number of motifs: 0#### $`10X`## number of barcodes: 4100## number of bins: 0## number of genes: 0## number of peaks: 0## number of motifs: 0
Step 3. Add cell-by-bin matrix
# 使用addBmatToSnap函数计算cell-by-bin计数矩阵并添加到snap对象中> x.sp.list = lapply(seq(x.sp.list), function(i){x.sp = addBmatToSnap(x.sp.list[[i]], bin.size=5000);x.sp})> x.sp.list## $snATAC## number of barcodes: 9646## number of bins: 545118## number of genes: 0## number of peaks: 0## number of motifs: 0#### $`10X`## number of barcodes: 4100## number of bins: 546206## number of genes: 0## number of peaks: 0## number of motifs: 0
可以看到,这两个snap对象中含有不同数目的bins,这是因为这两个数据集使用的参考基因组有细微的差异。
Step 4. Combine snap objects
接下来,我们将这个数据集进行合并。
To combine these two snap objects, common bins are selected.
# 选择两个数据集共有的bins> bin.shared = Reduce(intersect, lapply(x.sp.list, function(x.sp) x.sp@feature$name));> x.sp.list <- lapply(x.sp.list, function(x.sp){idy = match(bin.shared, x.sp@feature$name);x.sp[,idy, mat="bmat"];})# 合并两个数据集> x.sp = Reduce(snapRbind, x.sp.list);> rm(x.sp.list); # free memory> gc();> table(x.sp@sample);## 10X snATAC## 4100 9646
Step 5. Binarize matrix
# 使用makeBinary函数将计数矩阵转换为二进制矩阵> x.sp = makeBinary(x.sp, mat="bmat");
Step 6. Filter bins
首先,我们将与ENCODE中blacklist区域重叠的bins进行过滤,以防止潜在的artifacts。
> system("wget http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/mm10-mouse/mm10.blacklist.bed.gz");> library(GenomicRanges);> black_list = read.table("mm10.blacklist.bed.gz");> black_list.gr = GRanges(black_list[,1],IRanges(black_list[,2], black_list[,3]));> idy = queryHits(findOverlaps(x.sp@feature, black_list.gr));> if(length(idy) > 0){x.sp = x.sp[,-idy, mat="bmat"]};> x.sp## number of barcodes: 13746## number of bins: 545015## number of genes: 0## number of peaks: 0## number of motifs: 0
接下来,我们将过滤掉那些不需要的染色体信息。
> chr.exclude = seqlevels(x.sp@feature)[grep("random|chrM", seqlevels(x.sp@feature))];> idy = grep(paste(chr.exclude, collapse="|"), x.sp@feature);> if(length(idy) > 0){x.sp = x.sp[,-idy, mat="bmat"]};> x.sp## number of barcodes: 13746## number of bins: 545011## number of genes: 0## number of peaks: 0## number of motifs: 0
第三,bins的覆盖率大致是服从对数正态分布的。我们将与不变特征(如管家基因的启动子)重叠的前5%的bins进行删除 。
> bin.cov = log10(Matrix::colSums(x.sp@bmat)+1);> bin.cutoff = quantile(bin.cov[bin.cov > 0], 0.95);> idy = which(bin.cov <= bin.cutoff & bin.cov > 0);> x.sp = x.sp[, idy, mat="bmat"];> x.sp## number of barcodes: 13746## number of bins: 479127## number of genes: 0## number of peaks: 0## number of motifs: 0
Step 7. Reduce dimensionality
我们使用diffusion maps的方法来计算landmark diffusion maps进行数据降维。首先,我们随机选择出10,000个细胞作为landmarks,然后将剩余的query细胞映射到diffusion maps embedding中。
> row.covs = log10(Matrix::rowSums(x.sp@bmat)+1);> row.covs.dens = density(x = row.covs,bw = 'nrd', adjust = 1);> sampling_prob = 1 / (approx(x = row.covs.dens$x, y = row.covs.dens$y, xout = row.covs)$y + .Machine$double.eps);> set.seed(1);> idx.landmark.ds = sort(sample(x = seq(nrow(x.sp)), size = 10000, prob = sampling_prob));> x.landmark.sp = x.sp[idx.landmark.ds,];> x.query.sp = x.sp[-idx.landmark.ds,];> x.landmark.sp = runDiffusionMaps(obj= x.landmark.sp,input.mat="bmat",num.eigs=50);> x.query.sp = runDiffusionMapsExtension(obj1=x.landmark.sp,obj2=x.query.sp,input.mat="bmat");> x.landmark.sp@metaData$landmark = 1;> x.query.sp@metaData$landmark = 0;> x.sp = snapRbind(x.landmark.sp, x.query.sp);## combine landmarks and query cells;> x.sp = x.sp[order(x.sp@sample),]; # IMPORTANT> rm(x.landmark.sp, x.query.sp); # free memory
Step 8. Determine significant components
> plotDimReductPW(
obj=x.sp,
eigs.dims=1:50,
point.size=0.3,
point.color="grey",
point.shape=19,
point.alpha=0.6,
down.sample=5000,
pdf.file.name=NULL,
pdf.height=7,
pdf.width=7
);

Step 9. Remove batch effect
> library(harmony);
# 使用runHarmony函数进行批次校正
> x.after.sp = runHarmony(
obj=x.sp,
eigs.dims=1:22,
meta_data=x.sp@sample # sample index
);
Step 10. Graph-based cluster
> x.after.sp = runKNN(
obj= x.after.sp,
eigs.dim=1:22,
k=15
);
> x.after.sp = runCluster(
obj=x.after.sp,
tmp.folder=tempdir(),
louvain.lib="R-igraph",
path.to.snaptools=NULL,
seed.use=10
);
> x.after.sp@metaData$cluster = x.after.sp@cluster;
Step 11. Visualization
> x.sp = runViz(
obj=x.sp,
tmp.folder=tempdir(),
dims=2,
eigs.dims=1:22,
method="Rtsne",
seed.use=10
);
> x.after.sp = runViz(
obj=x.after.sp,
tmp.folder=tempdir(),
dims=2,
eigs.dims=1:22,
method="Rtsne",
seed.use=10
);
> par(mfrow = c(2, 3));
> plotViz(
obj=x.sp,
method="tsne",
main="Before Harmony",
point.color=x.sp@sample,
point.size=0.1,
text.add= FALSE,
down.sample=10000,
legend.add=TRUE
);
> plotViz(
obj=x.after.sp,
method="tsne",
main="After Harmony",
point.color=x.sp@sample,
point.size=0.1,
text.add=FALSE,
down.sample=10000,
legend.add=TRUE
);
> plotViz(
obj=x.after.sp,
method="tsne",
main="Cluster",
point.color=x.after.sp@cluster,
point.size=0.1,
text.add=TRUE,
text.size=1,
text.color="black",
text.halo.add=TRUE,
text.halo.color="white",
text.halo.width=0.2,
down.sample=10000,
legend.add=FALSE
);
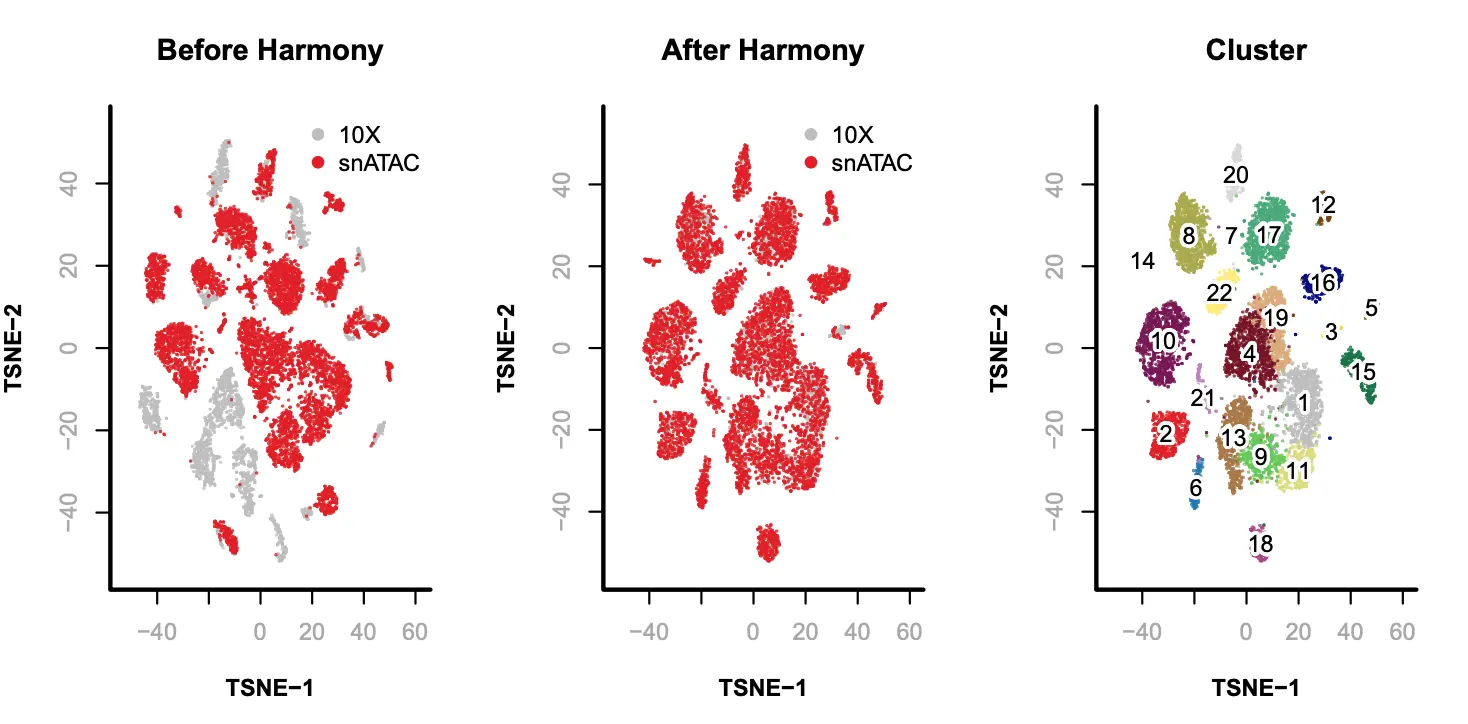
参考来源:https://gitee.com/booew/SnapATAC/blob/master/examples/10X_snATAC/README.md

若有收获,就点个赞吧
0 人点赞
