简介
immunarch包是一个主要面向于医学科学家和生物信息学家们设计的,专门针对 T 细胞受体 (TCR) 和 B 细胞受体 (BCR) 免疫组库数据进行分析的R包。使用immunarch包,使得免疫组库测序数据的分析变的尽可能轻松,并帮助您专注于研究而非编程。
为什么使用immunarch?
- 可以分析处理任何类型的免疫组库数据: single-cell、bulk、data tables、databases — 随心所欲。
- 以社区为核心: 在全球近 30,000 名研究人员和医学科学家组成的社区中提出问题、分享知识并茁壮成长。辉瑞、诺华、再生元、斯坦福、加州大学旧金山分校和麻省理工学院都信任我们。
- 一行代码一张图: 用8 行代码写一篇完整的博士论文或用 5-10 行
immunarch代码复制出几乎所有出版物中的图表。 - 站在科学的前沿: 我们会定期更新
immunarch最新的方法。让我们知道你需要什么! - 自动检测和解析 所有流行的免疫测序格式:从MiXCR和ImmunoSEQ到10XGenomics和ArcherDX。
特征
- 快速简便地操作免疫组库数据:
- 使得免疫组库数据分析变得简单:
- 大多数分析方法都包含在几个具有明确命名的主要函数中——不用再去记住数十个名称晦涩的函数。详情见链接;
- 克隆库重叠分析(Repertoire overlap analysis )(常用指标包括重叠系数、Jaccard 指数和 Morisita 重叠指数)。教程可在此处获得;
- TCR/BCR基因使用估计(Gene usage estimation )(相关性、Jensen-Shannon Divergence、聚类)。教程可以在这里找到;
- 克隆多样性评价(Diversity evaluation)(生态多样性指数、基尼指数、逆辛普森指数、稀疏分析)。教程可以在这里找到;
- 跨时间点克隆型追踪(Tracking of clonotypes across time points),广泛用于疫苗接种和癌症免疫学领域。教程可以在这里找到;
- Kmer分布测量和统计(Kmer distribution measures and statistics)。教程可以在这里找到;
- 即将发布的下一个版本:CDR3 氨基酸序列理化特性评估、突变网络。
Immunarch包的安装
可以直接在CRAN上安装,或者通过GitHub安装最新或的开发版本。
Latest release on CRAN
install.packages("immunarch")
Latest release on GitHub
install.packages("devtools") # skip this if you already installed devtoolsdevtools::install_github("immunomind/immunarch")
Latest pre-release on GitHub
install.packages("devtools") # skip this if you already installed devtoolsdevtools::install_github("immunomind/immunarch", ref="dev")
快速开始
接下来,我们将使用immunarch包内置的测试数据集进行演示 TCR 或 BCR的数据分析工作流程。
1) 加载包和示例数据
# 加载所需的包和数据集library(immunarch) # Load the package into Rdata(immdata) # Load the test dataset# 查看示例数据names(immdata)#[1] "data" "meta"names(immdata$data)# [1] "A2-i129" "A2-i131" "A2-i133" "A2-i132" "A4-i191" "A4-i192" "MS1" "MS2" "MS3"# [10] "MS4" "MS5" "MS6"head(immdata$data$`A2-i129`)## A tibble: 6 x 15# Clones Proportion CDR3.nt CDR3.aa V.name D.name J.name V.end D.start D.end J.start VJ.ins# <dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <int> <int> <int> <int> <dbl>#1 173 0.0204 TGCGCC… CASSQE… TRBV4… TRBD1 TRBJ2… 16 18 26 31 -1#2 163 0.0192 TGCGCC… CASSYR… TRBV4… TRBD1 TRBJ2… 11 13 18 22 -1#3 66 0.00776 TGTGCC… CATSTN… TRBV15 TRBD1 TRBJ2… 11 16 22 34 -1#4 54 0.00635 TGTGCC… CATSIG… TRBV15 TRBD2 TRBJ2… 11 19 25 26 -1#5 48 0.00565 TGTGCC… CASSPW… TRBV27 TRBD1 TRBJ1… 11 16 23 31 -1#6 48 0.00565 TGCGCC… CASQGD… TRBV4… TRBD1 TRBJ1… 8 13 19 23 -1# … with 3 more variables: VD.ins <dbl>, DJ.ins <dbl>, Sequence <lgl>names(immdata$data$`A2-i129`)# [1] "Clones" "Proportion" "CDR3.nt" "CDR3.aa" "V.name" "D.name" "J.name"# [8] "V.end" "D.start" "D.end" "J.start" "VJ.ins" "VD.ins" "DJ.ins"# [15] "Sequence"head(immdata$meta)## A tibble: 6 x 6# Sample ID Sex Age Status Lane# <chr> <chr> <chr> <dbl> <chr> <chr>#1 A2-i129 C1 M 11 C A#2 A2-i131 C2 M 9 C A#3 A2-i133 C4 M 16 C A#4 A2-i132 C3 F 6 C A#5 A4-i191 C8 F 22 C B#6 A4-i192 C9 F 24 C B
该示例数据集主要由两部分组成,其中data部分存储了12个样本的TCR或BCR注释信息,meta部分存储了这12个样本的metadata注释信息。
其中,data部分存储的每个样本的TCR或BCR注释信息包括以下条目:
- “
Clones”——条形码(events, UMIs)或reads的计数或数量; - “
Proportion”——条形码(events, UMIs)或reads的比例; - “
CDR3.nt”——CDR3核苷酸序列; - “
CDR3.aa”——CDR3氨基酸序列; - “
V.name”——对齐的Variable 基因片段的名称; - “
D.name”——对齐的Diversity 基因片段或 NA 的名称; - “
J.name”——对齐的Joining基因片段的名称; - “
V.end”——对齐的 V 基因片段的最后位置(基于 1); - “
D.start”——对齐的 D 基因片段的 D’5 末端的位置(基于 1); - “
D.end”——对齐的 D 基因片段的 D’3 末端的位置(基于 1); - “
J.start”——对齐的 J 基因片段的第一个位置(基于 1); - “
VJ.ins”——VJ 连接处插入核苷酸(N-核苷酸)的数量(-1 用于 VDJ 重组的受体); - “
VD.ins”——VD 连接处插入核苷酸(N-核苷酸)的数量(-1 用于 VJ 重组的受体); - “
DJ.ins”——DJ 连接处插入核苷酸(N-核苷酸)的数量(-1 用于 VJ 重组的受体); - “
Sequence”——完整的核苷酸序列。
2) 计算和可视化基本统计数据
# 可视化CDR3区的序列长度分布
repExplore(immdata$data, "lens") %>% vis() # Visualise the length distribution of CDR3
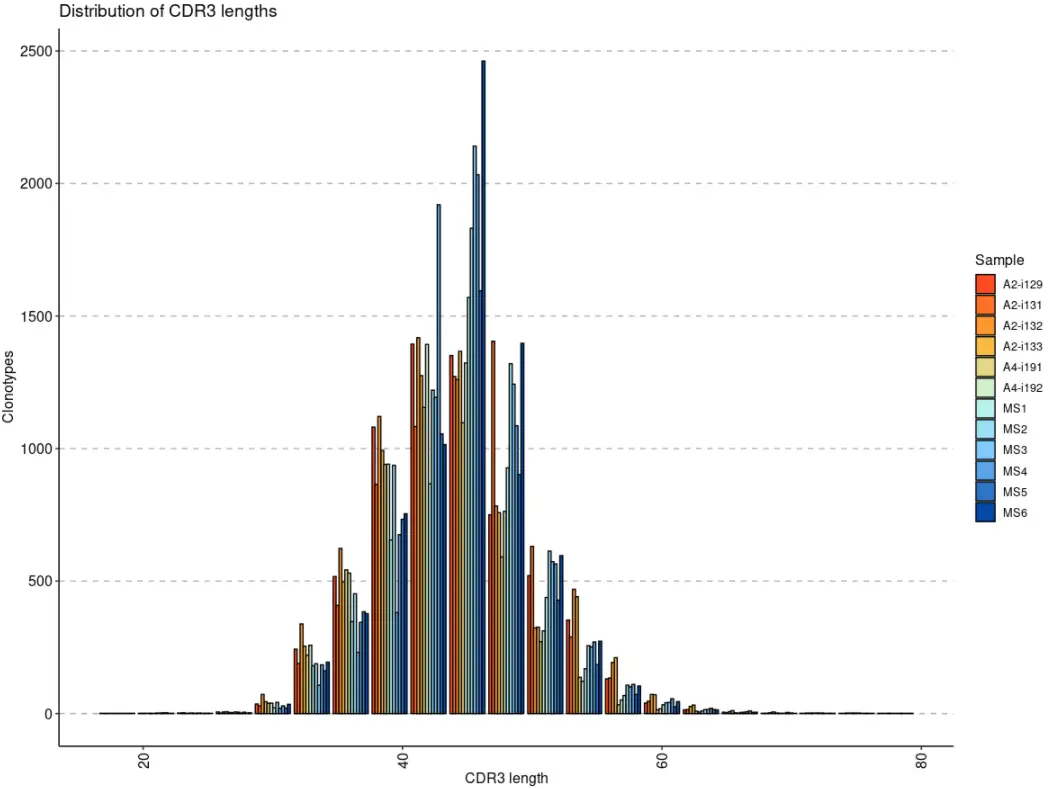
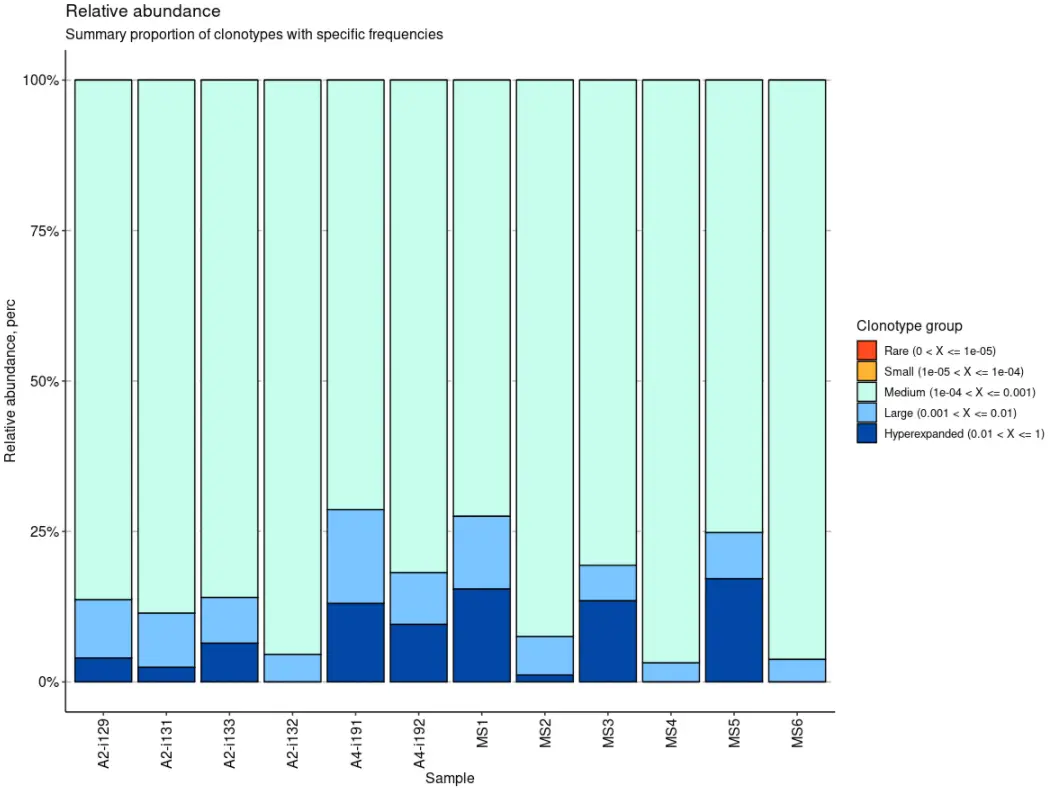
# 可视化克隆型的相对丰度
repClonality(immdata$data, "homeo") %>% vis() # Visualise the relative abundance of clonotypes
3) 探索和比较 T 细胞和 B 细胞库
# 构建不同克隆型之间共享的重叠克隆型热图
repOverlap(immdata$data) %>% vis() # Build the heatmap of public clonotypes shared between repertoires
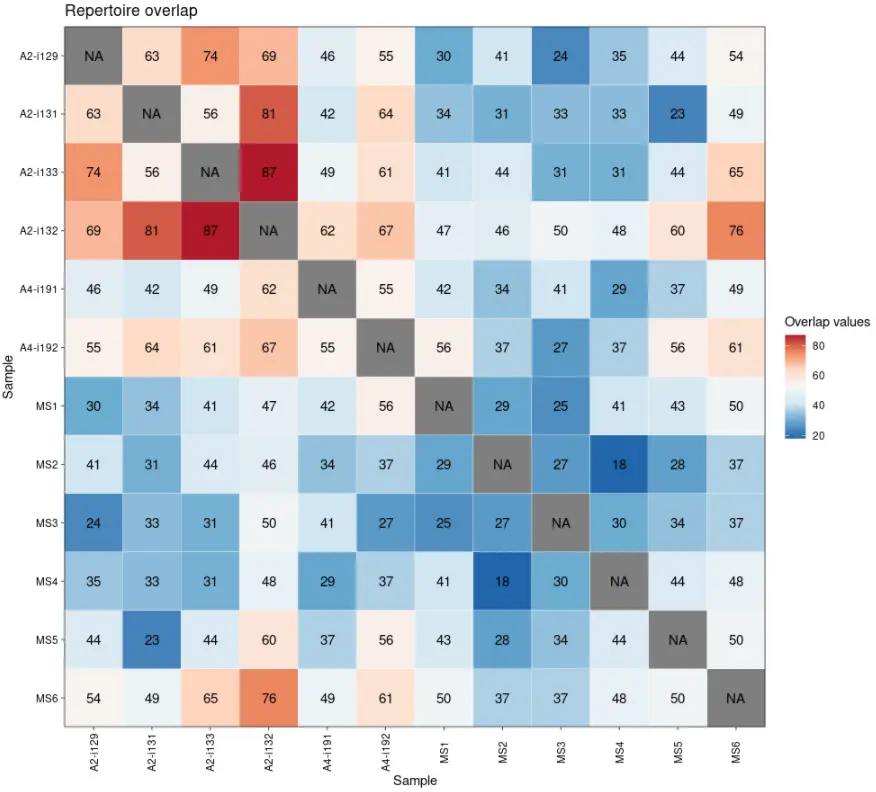
# 可视化第一个克隆型的V基因分布图
geneUsage(immdata$data[[1]]) %>% vis() # Visualise the V-gene distribution for the first repertoire
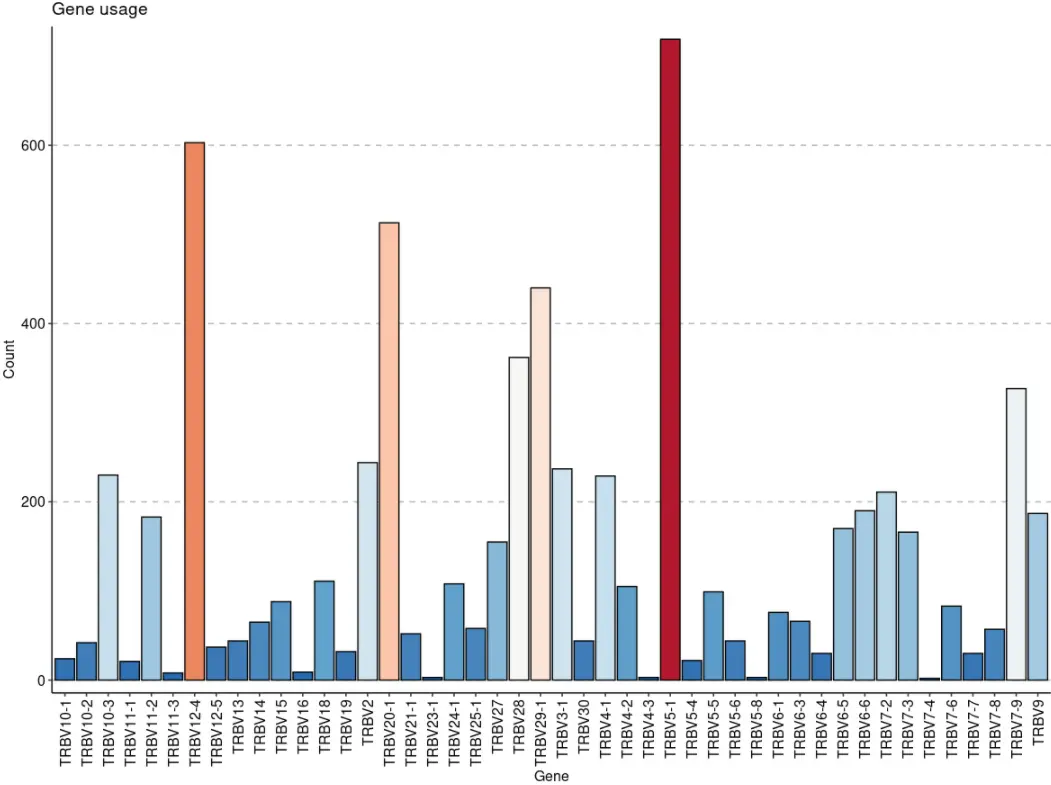
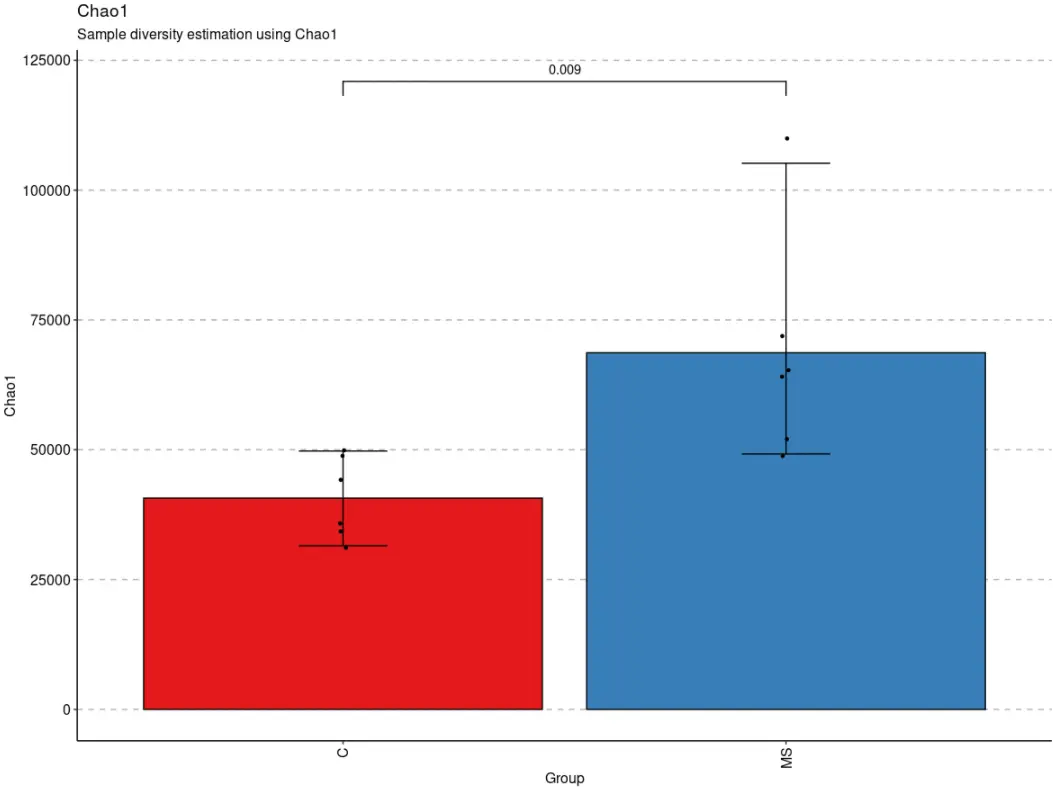
# 可视化克隆型的多样性
repDiversity(immdata$data) %>% vis(.by = "Status", .meta = immdata$meta) # Visualise the Chao1 diversity of repertoires, grouped by the patient status
使用自己的数据
immunarch包可以使用repLoad函数加载读取自己的数据集进行后续的分析,使用repSave函数保存分析的结果,repLoad函数可以自动检测输入文件的格式,我们可以通过?repLoad查看更详细的数据导入信息。
目前,immunarch包可以支持以下免疫组库数据的格式:
"immunarch"- 当前的软件工具,以防您忘记它:)"immunoseq"- http://www.adaptivebiotech.com/immunoseq"mitcr"- https://github.com/milaboratory/mitcr"mixcr"- https://github.com/milaboratory/mixcr"migec"- http://migec.readthedocs.io/en/latest/"migmap"- https://github.com/mikessh/migmap"tcr"- https://imminfo.github.io/tcr/"vdjtools"- https://vdjtools-doc.readthedocs.io/en/master/"imgt"- http://www.imgt.org/HighV-QUEST/"airr"- http://docs.airr-community.org/en/latest/datarep/overview.html"10x"- https://support.10xgenomics.com/single-cell-vdj/software/pipelines/latest/output/annotation"archer"- ArcherDX clonotype tables. https://archerdx.com/immunology/
library(immunarch) # Load the package into R
immdata <- repLoad("path/to/your/data") # Replace it with the path to your data. Immunarch automatically detects the file format.
参考来源:https://immunarch.com/index.html

若有收获,就点个赞吧
0 人点赞
