第二节:数据降维可视化实战
加载所需的R包和质控过滤后的数据
首先,我们加载分析所需的R包和上一节中质控过滤后的数据集。
# 加载所需的R包suppressPackageStartupMessages({library(Seurat)library(cowplot)library(ggplot2)library(scran)})# 加载质控过滤后的数据alldata <- readRDS("data/results/seurat_covid_qc.rds")# 查看数据信息alldata#An object of class Seurat#18121 features across 5532 samples within 1 assay#Active assay: RNA (18121 features, 2000 variable features)# 2 dimensional reductions calculated: pca, umap
特征选择筛选高变基因
接下来,我们需要筛选出数据集中细胞之间高度可变的特征/基因,用于后续的降维分析,以区分不同的细胞类型。
alldata <- FindVariableFeatures(alldata, selection.method = "vst", assay = "RNA"nfeatures = 2000, verbose = FALSE)top20 <- head(VariableFeatures(alldata), 20)head(top20)# [1] "IGLC3" "CXCL10" "IFI27" "PTGDS" "LYPD2" "STMN1"# 高变基因可视化LabelPoints(plot = VariableFeaturePlot(alldata), points = top20, repel = TRUE)
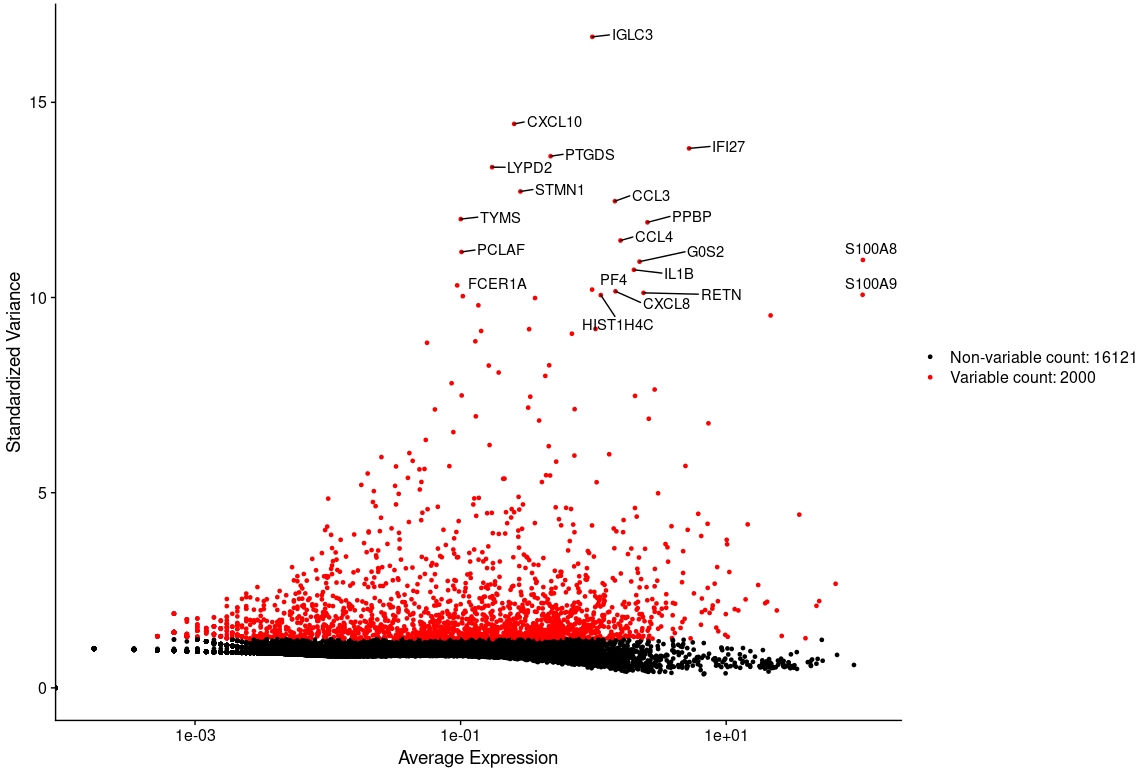
Z-score数据归一化
接下来,我们对数据进行z-score归一化处理。由于每个基因具有不同的表达水平,这意味着具有较高表达值的基因将会具有较高的变异,这些变异也更容易被PCA捕获。因此,在进行PCA降维之前,我们需要以某种方式赋予每个基因类似的权重(请参见下文)。常见的做法是在进行PCA之前对每个基因进行中心化(centering)和缩放(scaling)。这种精确的缩放比例称为Z-score归一化,对于PCA,聚类和绘制热图都是非常有用的。
此外,我们还可以使用回归的方法来从数据集中删除任何不想要的变异源,例如cell cycle,sequencing depth,percent mitocondria。我们可以通过将这些参数作为模型中的协变量进行广义线性回归来实现,然后将模型的残差作为“回归数据”。
alldata <- ScaleData(alldata, vars.to.regress = c("percent_mito", "nFeature_RNA"),assay = "RNA")
PCA线性降维
# 使用RunPCA函数进行PCA线性降维alldata <- RunPCA(alldata, npcs = 50, verbose = F)
然后,我们绘制前几个主成分。
plot_grid(ncol = 3, DimPlot(alldata, reduction = "pca", group.by = "orig.ident", dims = 1:2),DimPlot(alldata, reduction = "pca", group.by = "orig.ident", dims = 3:4),DimPlot(alldata, reduction = "pca", group.by = "orig.ident", dims = 5:6))

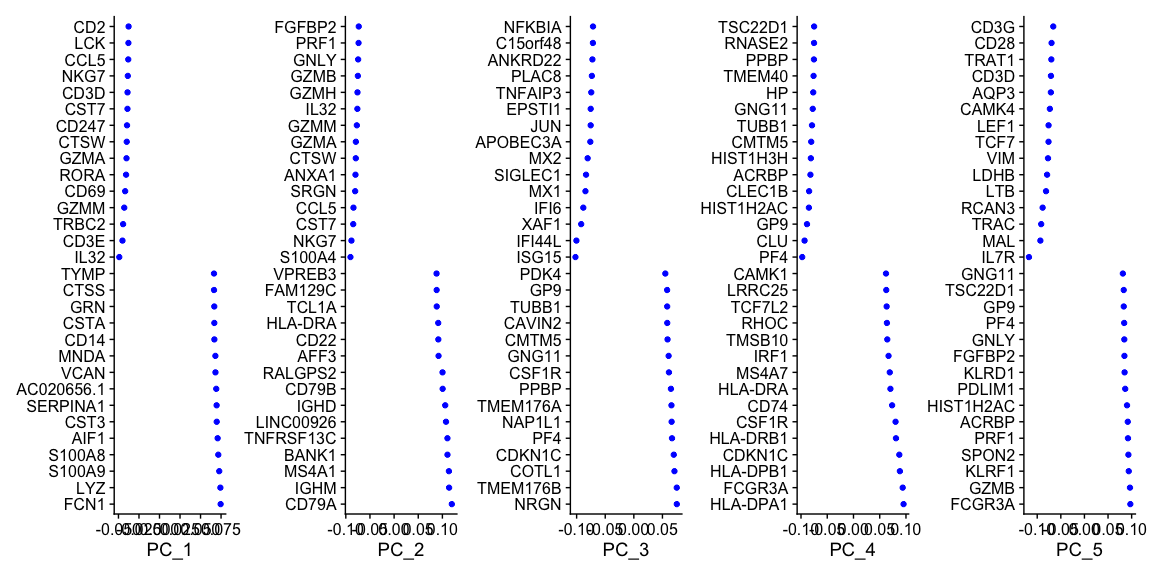
为了确定哪些基因或元数据对每个PC的贡献最大,我们可以使用VizDimLoadings函数查看相应基因对每个PC的贡献信息。
VizDimLoadings(alldata, dims = 1:5, reduction = "pca", ncol = 5, balanced = T)
我们还可以使用ElbowPlot函数绘制每个PC解释的方差量,以确定后续所要使用的PC数。
ElbowPlot(alldata, reduction = "pca", ndims = 50)

如图所示,我们可以看到排名前10位的PC保留了大量变异信息,而其他的PC所包含的信息则较少。但是,仍然建议使用更多的PC,因为它们可能包含有关稀有细胞类型(例如此数据集中的血小板和DC)的信息。
t-SNE降维可视化
接下来,我们进行t-SNE降维可视化处理。
# 使用RunTSNE函数进行t-SNE降维可视化alldata <- RunTSNE(alldata, reduction = "pca", dims = 1:30,perplexity = 30, max_iter = 1000,theta = 0.5, eta = 200, num_threads = 0)# see ?Rtsne and ?RunTSNE for more info
现在,我们可以绘制t-SNE降维后的数据,可以清楚地看到数据集中存在一定的批次效应。
DimPlot(alldata, reduction = "tsne", group.by = "orig.ident")
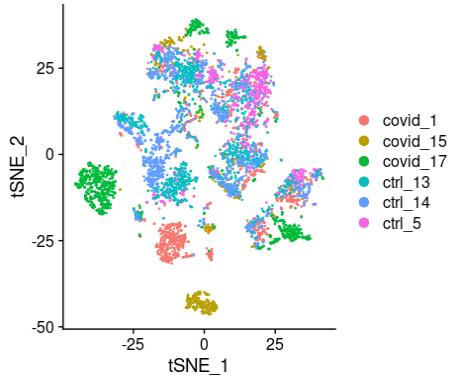
UMAP降维可视化
alldata <- RunUMAP(alldata, reduction = "pca", dims = 1:30,n.components = 2, n.neighbors = 30,n.epochs = 200, min.dist = 0.3,learning.rate = 1, spread = 1)# see ?RunUMAP for more infoDimPlot(alldata, reduction = "umap", group.by = "orig.ident")
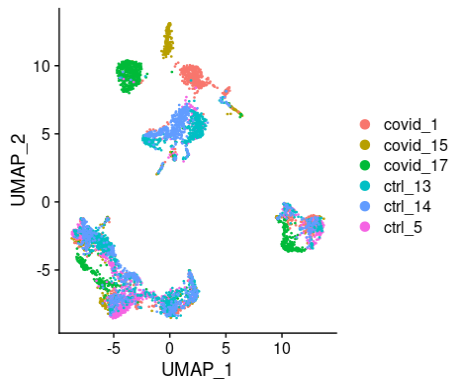
Task:测试设置不同的参数
UMAP降维的另一个有用之处是,它不受数据维数的限制(与tSNE不同),我们可以简单地改变n.components参数的大小。
# we can add in additional reductions, by defulat they are named 'pca', 'umap',# 'tsne' etc. But we can specify alternative names with reduction.namealldata <- RunUMAP(alldata, reduction.name = "UMAP10_on_PCA", reduction = "pca",dims = 1:30, n.components = 10, n.neighbors = 30, n.epochs = 200,min.dist = 0.3, learning.rate = 1, spread = 1)# see ?RunUMAP for more info
现在,我们可以绘制UMAP降维后的结果。尽管不像tSNE那样明显,但我们仍然可以看到数据中不同批次的影响很大。
plot_grid(ncol = 3, DimPlot(alldata, reduction = "umap", group.by = "orig.ident") + ggplot2::ggtitle(label = "UMAP_on_PCA"),DimPlot(alldata, reduction = "UMAP10_on_PCA", group.by = "orig.ident", dims = 1:2) + ggplot2::ggtitle(label = "UMAP10_on_PCA"),DimPlot(alldata, reduction = "UMAP10_on_PCA", group.by = "orig.ident", dims = 3:4) + ggplot2::ggtitle(label = "UMAP10_on_PCA"))
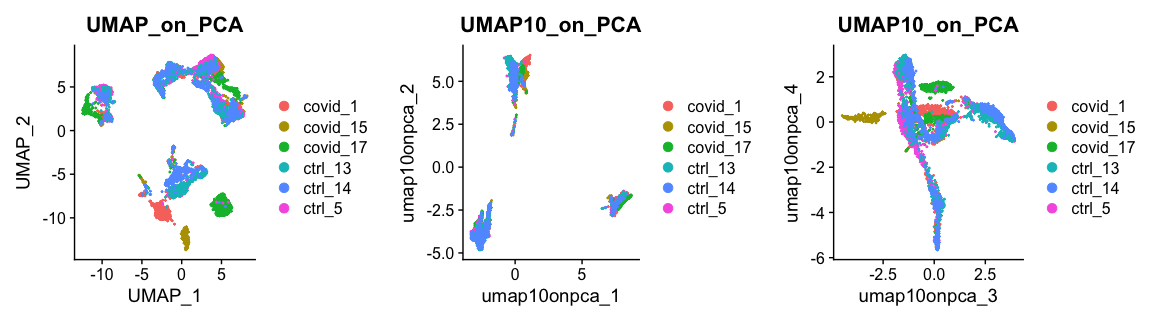
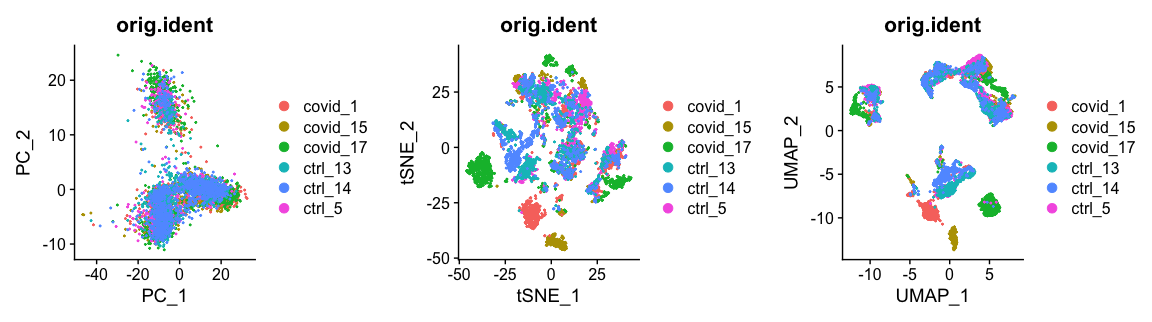
现在,我们可以并排绘制PCA,UMAP和tSNE降维的结果进行比较。在这里,我们可以得出结论,我们的数据集中存在一定的批处理效应,需要在进行后续聚类和差异基因表达分析之前对其进行校正。
plot_grid(ncol = 3, DimPlot(alldata, reduction = "pca", group.by = "orig.ident"),DimPlot(alldata, reduction = "tsne", group.by = "orig.ident"),DimPlot(alldata, reduction = "umap", group.by = "orig.ident"))
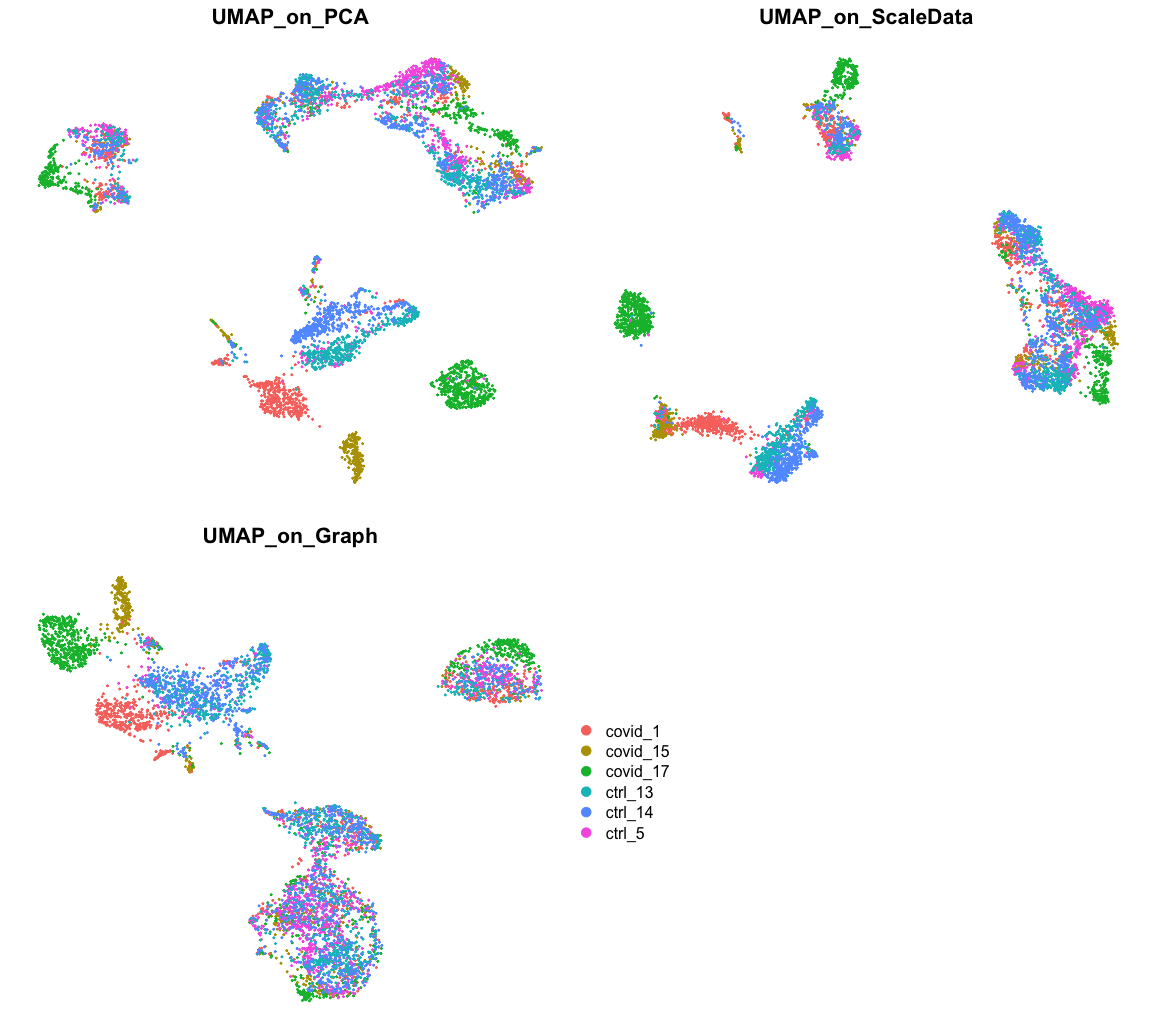
使用ScaledData和graphs进行数据降维
尽管在PCA线性降维后进行第二次降维(即tSNE或UMAP)将是一种标准方法(因为它允许更高的计算效率),但实际上这些选择是可变的。下面,我们将展示其他两个常见的做法,例如直接在缩放数据(用于PCA)上运行,或在根据缩放数据构建的图聚类上运行。从现在开始,我们将仅在UMAP上可视化降维的结果,但对于t-SNE同样如此。
使用ScaledData进行UMAP可视化
要对scaling后的数据进行t-SNE或UMAP降维可视化,首先需要选择要使用的变量数目。这是因为,那些有助于细胞类型分离的维数将最终掩盖这些差异。另一个原因是因为用所有的基因/特征一起运行也将花费更长的时间,或者可能在计算上不可行。因此,我们将使用高度可变的基因的缩放数据。
# 指定features参数设置所要使用的变异基因alldata <- RunUMAP(alldata, reduction.name = "UMAP_on_ScaleData",features = alldata@assays$RNA@var.features, assay = "RNA",n.components = 2, n.neighbors = 30, n.epochs = 200,min.dist = 0.3, learning.rate = 1, spread = 1)
使用图聚类进行UMAP可视化
要在图聚类上进行t-SNE或UMAP降维可视化,我们首先需要根据数据构建图形。实际上,t-SNE和UMAP都首先使用指定的距离矩阵从数据中构建图形,然后优化嵌入。由于图形只是一个矩阵,其中包含不同细胞之间的距离,因此,我们可以使用所需的任何其他距离度量方法来运行UMAP或tSNE。Euclidean和Correlation距离通常是最常用的距离度量。
# Build Graphalldata <- FindNeighbors(alldata, reduction = "pca", graph.name = "SNN",assay = "RNA", k.param = 20,features = alldata@assays$RNA@var.features)# Run UMAP on a graphalldata <- RunUMAP(alldata, reduction.name = "UMAP_on_Graph", graph = "SNN", assay = "RNA")
现在,我们可以在PCA,ScaledData和Graph上进行比较,绘制UMAP可视化图。
p1 <- DimPlot(alldata, reduction = "umap", group.by = "orig.ident") + ggplot2::ggtitle(label = "UMAP_on_PCA")p2 <- DimPlot(alldata, reduction = "UMAP_on_ScaleData", group.by = "orig.ident") + ggplot2::ggtitle(label = "UMAP_on_ScaleData")p3 <- DimPlot(alldata, reduction = "UMAP_on_Graph", group.by = "orig.ident") + ggplot2::ggtitle(label = "UMAP_on_Graph")leg <- get_legend(p1)gridExtra::grid.arrange(gridExtra::arrangeGrob(p1 + NoLegend() + NoAxes(), p2 + NoLegend() +NoAxes(), p3 + NoLegend() + NoAxes(), leg, nrow = 2), ncol = 1, widths = c(1))
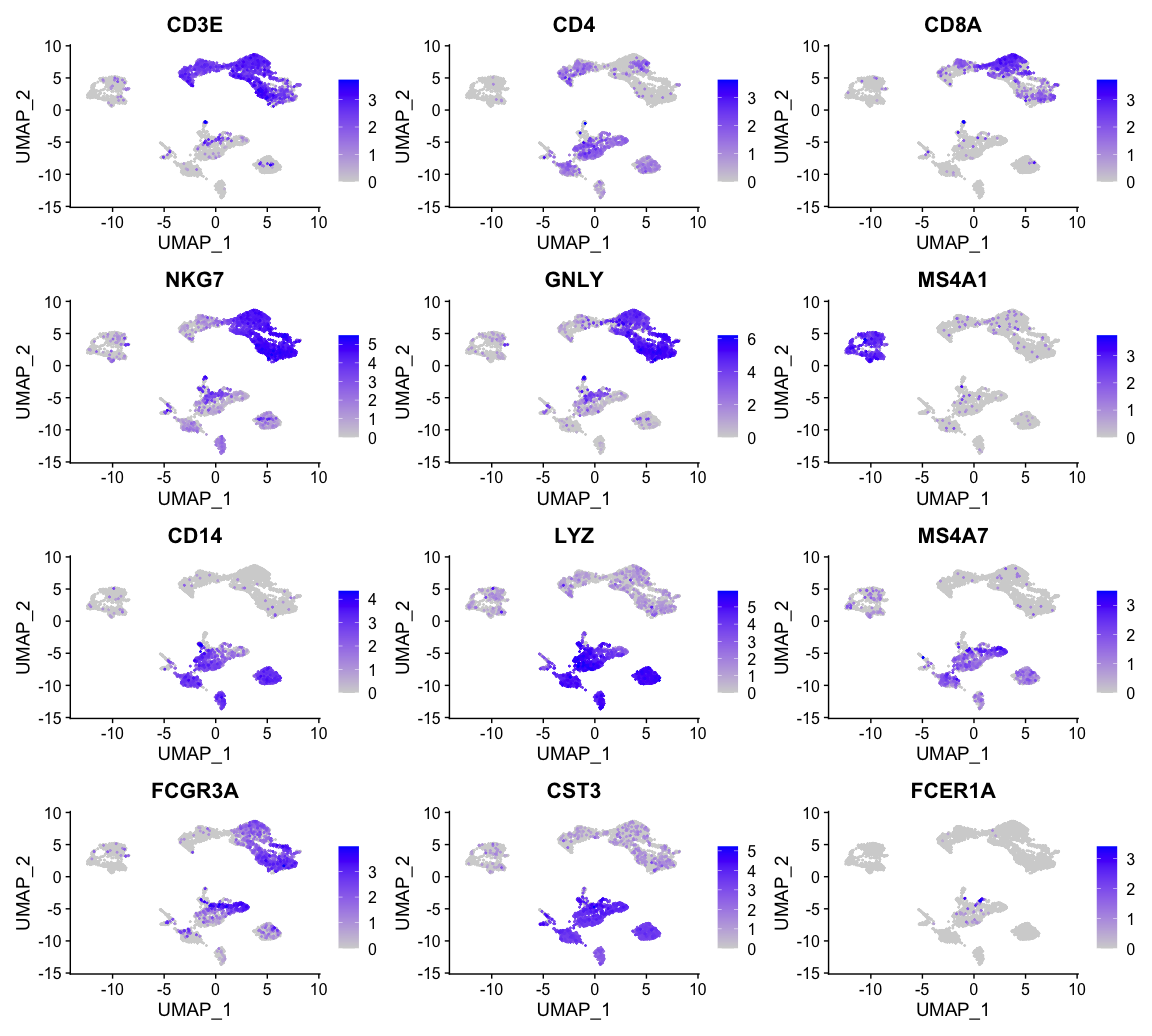
可视化感兴趣基因的表达
接下来,我们对一些不同细胞类型的marker基因进行可视化展示。
| Markers | Cell Type |
|---|---|
| CD3E | T cells |
| CD3E CD4 | CD4+ T cells |
| CD3E CD8A | CD8+ T cells |
| GNLY, NKG7 | NK cells |
| MS4A1 | B cells |
| CD14, LYZ, CST3, MS4A7 | CD14+ Monocytes |
| FCGR3A, LYZ, CST3, MS4A7 | FCGR3A+ Monocytes |
| FCER1A, CST3 | DCs |
myfeatures <- c("CD3E", "CD4", "CD8A", "NKG7", "GNLY", "MS4A1","CD14", "LYZ", "MS4A7", "FCGR3A", "CST3", "FCER1A")# 使用FeaturePlot函数进行可视化展示FeaturePlot(alldata, reduction = "umap", dims = 1:2,features = myfeatures, ncol = 3, order = T)
保存降维可视化后的数据
saveRDS(alldata, "data/results/covid_qc_dr.rds")
sessionInfo()
## R version 4.0.3 (2020-10-10)## Platform: x86_64-apple-darwin13.4.0 (64-bit)## Running under: macOS Catalina 10.15.5#### Matrix products: default## BLAS/LAPACK: /Users/paulo.czarnewski/.conda/envs/scRNAseq2021/lib/libopenblasp-r0.3.12.dylib#### locale:## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8#### attached base packages:## [1] parallel stats4 grid stats graphics grDevices utils## [8] datasets methods base#### other attached packages:## [1] scran_1.18.0 SingleCellExperiment_1.12.0## [3] SummarizedExperiment_1.20.0 Biobase_2.50.0## [5] GenomicRanges_1.42.0 GenomeInfoDb_1.26.0## [7] IRanges_2.24.0 S4Vectors_0.28.0## [9] BiocGenerics_0.36.0 MatrixGenerics_1.2.0## [11] matrixStats_0.57.0 ggplot2_3.3.3## [13] cowplot_1.1.1 KernSmooth_2.23-18## [15] fields_11.6 spam_2.6-0## [17] dotCall64_1.0-0 DoubletFinder_2.0.3## [19] Matrix_1.3-2 Seurat_3.2.3## [21] RJSONIO_1.3-1.4 optparse_1.6.6#### loaded via a namespace (and not attached):## [1] plyr_1.8.6 igraph_1.2.6## [3] lazyeval_0.2.2 splines_4.0.3## [5] BiocParallel_1.24.0 listenv_0.8.0## [7] scattermore_0.7 digest_0.6.27## [9] htmltools_0.5.1 magrittr_2.0.1## [11] tensor_1.5 cluster_2.1.0## [13] ROCR_1.0-11 limma_3.46.0## [15] remotes_2.2.0 globals_0.14.0## [17] colorspace_2.0-0 ggrepel_0.9.1## [19] xfun_0.20 dplyr_1.0.3## [21] crayon_1.3.4 RCurl_1.98-1.2## [23] jsonlite_1.7.2 spatstat_1.64-1## [25] spatstat.data_1.7-0 survival_3.2-7## [27] zoo_1.8-8 glue_1.4.2## [29] polyclip_1.10-0 gtable_0.3.0## [31] zlibbioc_1.36.0 XVector_0.30.0## [33] leiden_0.3.6 DelayedArray_0.16.0## [35] BiocSingular_1.6.0 future.apply_1.7.0## [37] maps_3.3.0 abind_1.4-5## [39] scales_1.1.1 edgeR_3.32.0## [41] DBI_1.1.1 miniUI_0.1.1.1## [43] Rcpp_1.0.6 viridisLite_0.3.0## [45] xtable_1.8-4 dqrng_0.2.1## [47] reticulate_1.18 bit_4.0.4## [49] rsvd_1.0.3 htmlwidgets_1.5.3## [51] httr_1.4.2 getopt_1.20.3## [53] RColorBrewer_1.1-2 ellipsis_0.3.1## [55] ica_1.0-2 scuttle_1.0.0## [57] pkgconfig_2.0.3 farver_2.0.3## [59] uwot_0.1.10 deldir_0.2-9## [61] locfit_1.5-9.4 tidyselect_1.1.0## [63] labeling_0.4.2 rlang_0.4.10## [65] reshape2_1.4.4 later_1.1.0.1## [67] munsell_0.5.0 tools_4.0.3## [69] generics_0.1.0 ggridges_0.5.3## [71] evaluate_0.14 stringr_1.4.0## [73] fastmap_1.0.1 yaml_2.2.1## [75] goftest_1.2-2 knitr_1.30## [77] bit64_4.0.5 fitdistrplus_1.1-3## [79] purrr_0.3.4 RANN_2.6.1## [81] sparseMatrixStats_1.2.0 pbapply_1.4-3## [83] future_1.21.0 nlme_3.1-151## [85] mime_0.9 formatR_1.7## [87] hdf5r_1.3.3 compiler_4.0.3## [89] plotly_4.9.3 curl_4.3## [91] png_0.1-7 spatstat.utils_1.20-2## [93] statmod_1.4.35 tibble_3.0.5## [95] stringi_1.5.3 RSpectra_0.16-0## [97] bluster_1.0.0 lattice_0.20-41## [99] vctrs_0.3.6 pillar_1.4.7## [101] lifecycle_0.2.0 lmtest_0.9-38## [103] BiocNeighbors_1.8.0 RcppAnnoy_0.0.18## [105] data.table_1.13.6 bitops_1.0-6## [107] irlba_2.3.3 httpuv_1.5.5## [109] patchwork_1.1.1 R6_2.5.0## [111] promises_1.1.1 gridExtra_2.3## [113] parallelly_1.23.0 codetools_0.2-18## [115] MASS_7.3-53 assertthat_0.2.1## [117] withr_2.4.0 sctransform_0.3.2## [119] GenomeInfoDbData_1.2.4 mgcv_1.8-33## [121] beachmat_2.6.0 rpart_4.1-15## [123] tidyr_1.1.2 DelayedMatrixStats_1.12.0## [125] rmarkdown_2.6 Rtsne_0.15## [127] shiny_1.5.0
参考来源:https://nbisweden.github.io/workshop-scRNAseq/labs/compiled/seurat/seurat_02_dim_reduction.html

若有收获,就点个赞吧
0 人点赞
