免疫受体数据库简介
具有关于免疫受体特异性的汇总信息的数据库提供了一种直接的方法来注释您的数据并找到与疾病相关的TCR或BCR受体。immunarch包中支持使用了最流行的 AIRR 数据库(VDJDB、McPAS-TCR 和 PIRD TBAdb)注释数据的工具。
数据库注释主要包括以下两步过程。首先,我们需要下载所需的数据库文件 - 完整的数据库文件或从数据库的 Web 界面获得的过滤数据。然后,再使用immunarch包中的函数来注释数据,并可视化注释结果。
下载注释数据库
VDJDB数据库
VDJDB数据库是一个已知抗原特异性的T细胞受体序列的精选数据库。该数据库基于 GitHub,可在此处获得:https : //github.com/antigenomics/vdjdb-db
参考文献: Shugay M et al. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Research 2017
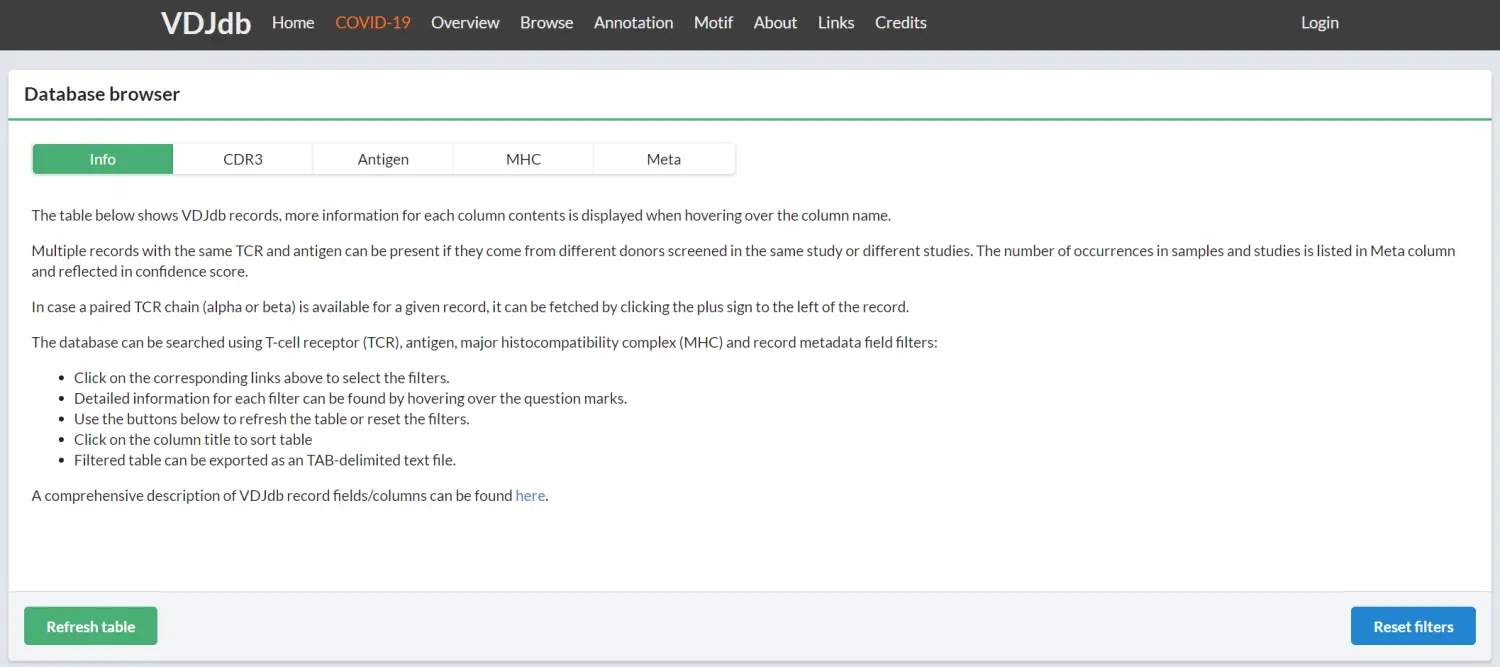
如何过滤和下载数据
在使用数据库之前,从总的数据库中过滤掉不相关的免疫受体信息是很有用的。例如,如果您分析人类 T 细胞 的TCR β 库,则没有必要保留来自其他物种的免疫受体以及非 TRB 数据。使用位于https://vdjdb.cdr3.net/search的VDJDB数据web界面过滤数据。选取过滤数据后按下“Refresh table”按钮,找到“Export”按钮并选择里面的“TSV”标签。您将下载过滤后的数据库文件,其名称类似于“SearchTable-2019-10-17 12_36_11.989.tsv”,可用于immunarch包中进行克隆型注释.
如何下载完整的VDJDB数据库
如果您在“CDR3”选项卡的“General”部分中设置了所有复选标记,则可以使用上面的方法下载完整数据库。但是,如果您想下载原始数据库文件,以下给出了下载VDJDB数据库和解包的复杂分步指南:
- 首先,您需要安装 JDK 8 - Java 开发工具包。如果您已经安装了它,请跳过此步骤。如果没有,只需为您的系统搜索正确的安装说明。
- 其次,您需要安装 Groovy——一种用于处理 VDJDB 的语言。转到此链接并根据您的系统下载发行版或 Windows 安装程序。对于 Windows 用户,最好的方法是下载 Windows 安装程序。对于 Linux 用户,最简单的方法是使用操作系统包管理器,例如 apt、dpkg、pacman 等。对于 Mac 用户,最无缝的方法是使用Homebrew。
- 通过此链接从 GitHub 下载 VDJDB 存储库:https : //github.com/antigenomics/vdjdb-db/archive/master.zip
- 解压缩存档并转到解压后的“vdjdb-db-master”文件夹。
- 转到“src”文件夹。
- 打开您的终端或控制台并执行以下命令:
groovy -cp . BuildDatabase.groovy --no2fix. - 经过一些处理后,数据库文件将位于“vdjdb-db-master”文件夹内的“database”文件夹中。您需要为
immunarch注释函数提供此文件的路径。
McPAS-TCR数据库
McPAS-TCR数据库是一个人工整理的病理相关的 T 细胞受体序列目录。该数据库位于http://friedmanlab.weizmann.ac.il/McPAS-TCR/
参考文献:Shugay M et al. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Research 2017
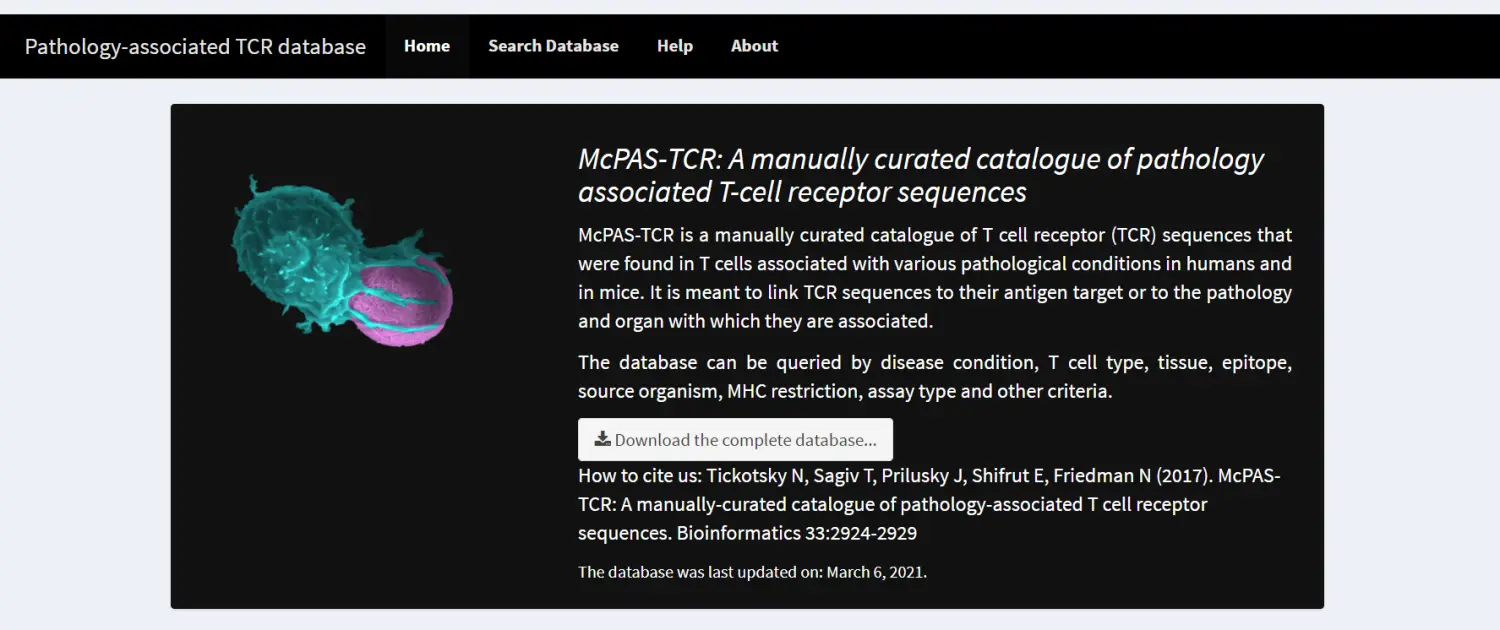
如何过滤和下载数据
McPAS-TCR数据库 Web 界面的过滤功能位于“Search Database”选项卡中。筛选完数据后,按“Download csv”按钮即可下载。下载的名为“McPAS-TCR_search.csv”的文件可用于immunarch包进行注释分析。
如何下载完整的McPAS-TCR
要下载完整的McPAS-TCR,您只需访问http://friedmanlab.weizmann.ac.il/McPAS-TCR/并在那里按“Download the complete database”按钮。请注意,有时您需要按两次或在新的浏览器选项卡中按它才能开始下载过程。
TBAdb数据库
TBAdb数据库是一个人工管理的针对特定抗原或疾病的 T 细胞受体 (TCR) 和 B 细胞受体 (BCR)数据库。该数据库包含三部分,即TCR-AB、TCR-GD和BCR。这三个部分旨在分别收集TCRA和TCRB、TCR-gamma和TCR-delta和BCR的序列和特异性信息。该数据库可在https://db.cngb.org/pird/tbadb/ 获得
参考文献: Tickotsky N, Sagiv T, Prilusky J, Shifrut E, Friedman N (2017). McPAS-TCR: A manually-curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics 33:2924-2929
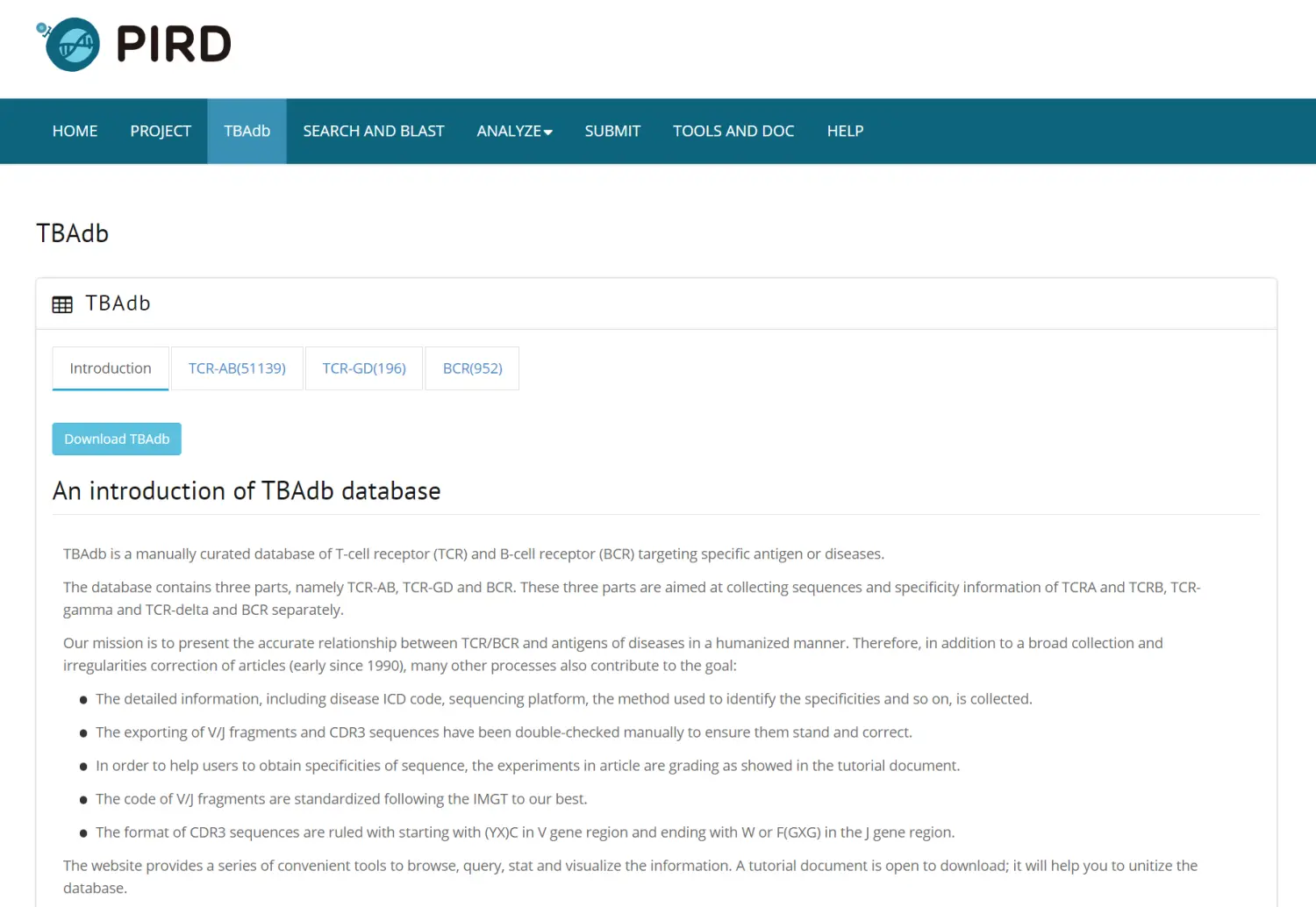
如何过滤和下载数据
目前无法从 TBAdb数据库 下载过滤后的数据。查询功能可在https://db.cngb.org/pird/query/的“TBAdb”选项卡中使用。
如何下载完整的 TBAdb数据库
要下载完整的TBAdb数据库,您需要访问https://db.cngb.org/pird/tbadb/并按“Download TBAdb”按钮。请注意,您应该同意许可协议才能下载数据库文件。
克隆型注释
下载好相应的注释数据库后,我们可以继续使用immunarch包进行克隆型注释分析。接下来,我们将使用巨细胞病毒 (CMV) 感染的克隆库进行注释分析。
使用 R 预处理数据库
首先,我们需要将数据库加载到 R 中,并从输入数据库中过滤掉非人类、非 TRB 和非 CMV的 数据。对于数据库,我们遵循与repLoad和vis函数相同的原理:函数dbLoad为所有支持的数据库的加载和基本查询提供了一个单一的接口。
出于演示目的,我们将处理以下每个受支持的数据库。
导入VDJDB数据库
按照上述说明下载 VDJDB 数据库。在下面的示例中,我们使用 URL 到数据库片段作为文件路径。在您自己的代码中,您需要提供本地数据库文件的路径,例如“/Users/yourname/Downloads/vdjdb-db-master/vdjdb.slim.txt”。不要使用下面的链接,因为它们仅用于测试目的,而不是实际的数据库!
请注意,从 Web 界面获取的 VDJDB 数据与从原始文件中获取的 VDJDB数据不同。
将 VDJDB数据加载到 R 的最基本方法:
vdjdb = dbLoad("https://gitlab.com/immunomind/immunarch/raw/dev-0.5.0/private/vdjdb.slim.txt.gz", "vdjdb")vdjdb## # A tibble: 61,049 x 19## gene cdr3 species antigen.epitope antigen.gene antigen.species complex.id## <chr> <chr> <chr> <chr> <chr> <chr> <dbl>## 1 TRB CASS… Macaca… STPESANL Tat SIV 0.## 2 TRB CASS… HomoSa… RLRAEAQVK EBNA3A EBV 1.93e 4## 3 TRB CASS… Macaca… TTPESANL Tat SIV 0.## 4 TRA CASN… HomoSa… GILGFVFTL M InfluenzaA 0.## 5 TRB CASS… MusMus… HGIRNASFI M45 MCMV 2.24e24## 6 TRB CSAS… HomoSa… KLGGALQAK IE1 CMV 8.58e 3## 7 TRA CAVL… HomoSa… GILGFVFTL M InfluenzaA 0.## 8 TRB CASS… HomoSa… KLGGALQAK IE1 CMV 3.44e 3## 9 TRB CAST… MusMus… SSYRRPVGI PB1 InfluenzaA 2.28e 4## 10 TRB CASS… HomoSa… NLVPMVATV pp65 CMV 0.## # … with 61,039 more rows, and 12 more variables: v.segm <chr>, j.segm <chr>,## # v.end <dbl>, j.start <dbl>, mhc.a <chr>, mhc.b <chr>, mhc.class <chr>,## # reference.id <chr>, vdjdb.score <dbl>, Species <chr>, Chain <chr>,## # Pathology <chr>
我们可以通过设置.species,.chain以及.pathology等参数进行过滤筛选出需要的信息:
vdjdb = dbLoad ( "https://gitlab.com/immunomind/immunarch/raw/dev-0.5.0/private/vdjdb.slim.txt.gz" , "vdjdb" , .species = "HomoSapiens" , .chain = "TRB" , .pathology = "CMV" )vdjdb## # A tibble: 18,039 x 19## gene cdr3 species antigen.epitope antigen.gene antigen.species complex.id## <chr> <chr> <chr> <chr> <chr> <chr> <dbl>## 1 TRB CSAS… HomoSa… KLGGALQAK IE1 CMV 8584## 2 TRB CASS… HomoSa… KLGGALQAK IE1 CMV 3445## 3 TRB CASS… HomoSa… NLVPMVATV pp65 CMV 0## 4 TRB CASS… HomoSa… KLGGALQAK IE1 CMV 19396## 5 TRB CASS… HomoSa… NLVPMVATV pp65 CMV 0## 6 TRB CAST… HomoSa… KLGGALQAK IE1 CMV 10972## 7 TRB CASS… HomoSa… KLGGALQAK IE1 CMV 6231## 8 TRB CASS… HomoSa… KLGGALQAK IE1 CMV 12587## 9 TRB CATS… HomoSa… KLGGALQAK IE1 CMV 13267## 10 TRB CASS… HomoSa… NLVPMVATV pp65 CMV 0## # … with 18,029 more rows, and 12 more variables: v.segm <chr>, j.segm <chr>,## # v.end <dbl>, j.start <dbl>, mhc.a <chr>, mhc.b <chr>, mhc.class <chr>,## # reference.id <chr>, vdjdb.score <dbl>, Species <chr>, Chain <chr>,## # Pathology <chr>
VDJDB search tables
vdjdb_st = dbLoad("https://gitlab.com/immunomind/immunarch/raw/dev-0.5.0/private/SearchTable-2019-10-17%2012_36_11.989.tsv.gz", "vdjdb-search", .species = "HomoSapiens", .chain = "TRB", .pathology = "CMV")vdjdb_st## # A tibble: 4,999 x 19## complex.id Gene CDR3 V J Species `MHC A` `MHC B` `MHC class`## <dbl> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>## 1 0 TRB CASS… TRBV… TRBJ… HomoSa… HLA-A*… B2M MHCI## 2 0 TRB CAWS… TRBV… TRBJ… HomoSa… HLA-A*… B2M MHCI## 3 0 TRB CASS… TRBV… TRBJ… HomoSa… HLA-A*… B2M MHCI## 4 0 TRB CASS… TRBV… TRBJ… HomoSa… HLA-B*… B2M MHCI## 5 0 TRB CASS… TRBV… TRBJ… HomoSa… HLA-B*… B2M MHCI## 6 0 TRB CASS… TRBV… TRBJ… HomoSa… HLA-B*… B2M MHCI## 7 0 TRB CASV… TRBV… TRBJ… HomoSa… HLA-B*… B2M MHCI## 8 0 TRB CASS… TRBV… TRBJ… HomoSa… HLA-B*… B2M MHCI## 9 0 TRB CASS… TRBV… TRBJ… HomoSa… HLA-B*… B2M MHCI## 10 0 TRB CASG… TRBV… TRBJ… HomoSa… HLA-B*… B2M MHCI## # … with 4,989 more rows, and 10 more variables: Epitope <chr>, `Epitope## # gene` <chr>, `Epitope species` <chr>, Reference <chr>, Method <chr>,## # Meta <chr>, CDR3fix <chr>, Score <dbl>, Chain <chr>, Pathology <chr>
导入McPAS-TCR数据库
mcpas = dbLoad("https://gitlab.com/immunomind/immunarch/raw/dev-0.5.0/private/McPAS-TCR.csv.gz", "mcpas", .species = "Human", .chain = "TRB", .pathology = "Cytomegalovirus (CMV)")mcpas## # A tibble: 2,723 x 29## CDR3.alpha.aa CDR3.beta.aa Species Category Pathology Pathology.Mesh.…## <chr> <chr> <chr> <chr> <chr> <chr>## 1 <NA> CASLAPGTTNE… Human Pathoge… Cytomega… D003586## 2 <NA> CASLQAGANEQF Human Pathoge… Cytomega… D003586## 3 <NA> CASLSGGGEQF Human Pathoge… Cytomega… D003586## 4 <NA> CASLVASGQET… Human Pathoge… Cytomega… D003586## 5 <NA> CASSHRDSGNT… Human Pathoge… Cytomega… D003586## 6 <NA> CASSSANYGYTF Human Pathoge… Cytomega… D003586## 7 <NA> CATSDPLTASY… Human Pathoge… Cytomega… D003586## 8 CARNTGNQF CACSLRSQGTD… Human Pathoge… Cytomega… D003586## 9 CAGNTGNQFYFG CASSAWDRSSG… Human Pathoge… Cytomega… D003586## 10 CAYPYNNNDMRF CASSELGGAGT… Human Pathoge… Cytomega… D003586## # … with 2,713 more rows, and 23 more variables:## # Additional.study.details <chr>, Antigen.identification.method <dbl>,## # NGS <chr>, Antigen.protein <chr>, Protein.ID <chr>, Epitope.peptide <chr>,## # Epitope.ID <dbl>, MHC <chr>, Tissue <chr>, T.Cell.Type <chr>,## # T.cell.characteristics <chr>, CDR3.alpha.nt <chr>, TRAV <chr>, TRAJ <chr>,## # TRBV <chr>, TRBD <chr>, TRBJ <chr>, Reconstructed.J.annotation <chr>,## # CDR3.beta.nt <chr>, Mouse.strain <chr>, PubMed.ID <dbl>, Remarks <chr>,## # Chain <chr>
导入TBAdb数据库
tbadb = dbLoad("https://gitlab.com/immunomind/immunarch/raw/dev-0.5.0/private/TBAdb.xlsx", "tbadb", .species = "Homo Sapiens", .chain = c("TRB", "TRA-TRB"), .pathology = "CMV")tbadb
免疫组库注释
在immunarch包中,我们可以使用dbAnnotate函数进行TCR或BCR克隆型的注释。在输入数据中,我们需要指定搜索的序列、要查找的数据库以及感兴趣的列,例如 CDR3 氨基酸序列或 V 基因片段名称列。
接下来,我们将使用immunarch包的内置数据进行测试。只需一行代码,就可以在输入数据和VDJDB数据库中找到所有匹配CDR3氨基酸序列的克隆型:
library(immunarch)data(immdata)dbAnnotate(immdata$data, vdjdb, "CDR3.aa", "cdr3")## CDR3.aa Samples A2-i129 A2-i131 A2-i133 A2-i132 A4-i191 A4-i192 MS1## 1: CASSLGETQYF 11 6 4 2 8 4 0 1## 2: CASSFQETQYF 9 3 2 2 4 2 0 1## 3: CASSQETQYF 9 5 2 1 2 3 2 0## 4: CASSSSYEQYF 9 1 0 0 1 2 2 1## 5: CASSLEGYEQYF 8 0 0 1 1 3 0 1## ---## 579: CSVGTGTYEQYF 1 0 0 0 0 1 0 0## 580: CSVQGGAYNEQFF 1 0 1 0 0 0 0 0## 581: CSVQGGSYNEQFF 1 0 1 0 0 0 0 0## 582: CSVVATNEKLFF 1 0 0 1 0 0 0 0## 583: CSVVGTGNTEAFF 1 0 0 0 0 0 0 0## MS2 MS3 MS4 MS5 MS6## 1: 3 1 2 5 1## 2: 1 0 4 0 2## 3: 1 0 0 4 1## 4: 1 0 1 1 3## 5: 0 1 1 1 1## ---## 579: 0 0 0 0 0## 580: 0 0 0 0 0## 581: 0 0 0 0 0## 582: 0 0 0 0 0## 583: 0 0 0 1 0
在输出结果中,“Sample”列指定了在数据库中找到克隆型的样本数。列中的其他数字对应于特定输入样本中克隆型的克隆计数。
在下一个示例中,我们将使用 CDR3 氨基酸序列和 V 基因片段在 McPAS-TCR 数据库中搜索与条件相关的克隆型:
dbAnnotate ( immdata $ data , mcpas , c ( "CDR3.aa" , "V.name" ), c ( "CDR3.beta.aa" , "TRBV" ))## CDR3.aa V.name Samples A2-i129 A2-i131 A2-i133 A2-i132 A4-i191## 1: CAISESYEQYF TRBV10-3 3 0 1 0 0 0## 2: CASSLAPGATNEKLFF TRBV7-6 3 0 0 0 0 0## 3: CASSLGENIQYF TRBV13 3 0 0 0 0 1## 4: CASSLGRETQYF TRBV28 3 0 0 0 0 0## 5: CSVGTGGTNEKLFF TRBV29-1 3 0 0 0 0 0## ---## 2123: KNPTAF TRBV19 1 0 0 0 0 0## 2124: LLGGQETQYF TRBV7-4 1 0 0 0 0 0## 2125: WASSFQGFTEAF TRBV28 1 0 0 0 0 0## 2126: WASSQALPYEQYF TRBV12-4 1 0 0 0 0 0## 2127: WASSQQTGTIGGYTF TRBV6-5 1 0 0 0 0 0## A4-i192 MS1 MS2 MS3 MS4 MS5 MS6## 1: 1 0 0 0 0 0 0## 2: 0 0 1 0 0 1 0## 3: 1 0 0 0 0 0 0## 4: 0 0 0 0 0 75 1## 5: 0 0 1 0 0 0 1## ---## 2123: 0 0 0 0 0 0 0## 2124: 0 0 0 0 0 0 0## 2125: 0 0 0 0 0 0 0## 2126: 0 0 0 0 0 0 0## 2127: 0 0 0 0 0 0 0
如果您想在数据库中搜索一组特定克隆型的序列,请使用它们创建一个数据框并将其作为一个数据库文件:
local_db = data.frame ( Seq = c ( "CASSDSSGGANEQFF" , "CSARLAGGQETQYF" ), V = c ( "TRBV6-4" , "TRBV20-1" ), stringsAsFactors = F )dbAnnotate ( immdata $ data , local_db , c ( "CDR3.aa" , "V.name" ), c ( "Seq" , "V" ))## CDR3.aa V.name Samples A2-i129 A2-i131 A2-i133 A2-i132 A4-i191## 1: CASSDSSGGANEQFF TRBV6-4 7 1 1 2 0 3## 2: CSARLAGGQETQYF TRBV20-1 6 1 3 0 1 0## A4-i192 MS1 MS2 MS3 MS4 MS5 MS6## 1: 0 0 0 2 0 0 12## 2: 0 0 0 1 0 0 1
高级过滤
immunarch包中提供了一个非常基本的查询界面,只允许按物种类型、链类型和病理类型进行过滤。要执行高级过滤,例如按抗原表位过滤,您需要使用 R。在大多数情况下,使用dplyr包过滤是最无缝的方式。以下是如何使用dplyr从 VDJDB 中过滤掉特定抗原表位的示例:
# Load the dplyr librarylibrary(dplyr)# Load the database with immunarchvdjdb = dbLoad("https://gitlab.com/immunomind/immunarch/raw/dev-0.5.0/private/vdjdb.slim.txt.gz", "vdjdb", .species = "HomoSapiens", .chain = "TRB", .pathology = "CMV")# Check which antigen epitopes are presented in the database#检查数据库中收录的抗原表位table(vdjdb$antigen.epitope)#### ARNLVPMVATVQGQN AYAQKIFKI CPSQEPMSIYVY CVETMCNEY DEEDAIAAY## 3 39 2 2 2## EDVPSGKLFMHVTLG EFFWDANDIY ELKRKMIYM ELRRKMMYM FPTKDVAL## 1 1 5 10 10## IPSINVHHY KLGGALQAK LSEFCRVLCCYVLEE MLNIPSINV NEGVKAAW## 93 12667 2 73 49## NLVPMVATV QIKVRVDMV QIKVRVKMV QYDPVAALF RPHERNGFTV## 4496 15 24 39 4## RPHERNGFTVL TPRVTGGGAM VLEETSVML VMAPRTLIL VTEHDTLLY## 22 207 14 1 202## YILEETSVM YSEHPTFTSQY## 3 53
# Filter out all non NLVPMVATV epitopes# 筛选出所有非NLVPMVATV表位vdjdb = vdjdb %>% filter(antigen.epitope == "NLVPMVATV")vdjdb## # A tibble: 4,496 x 19## gene cdr3 species antigen.epitope antigen.gene antigen.species complex.id## <chr> <chr> <chr> <chr> <chr> <chr> <dbl>## 1 TRB CASS… HomoSa… NLVPMVATV pp65 CMV 0## 2 TRB CASS… HomoSa… NLVPMVATV pp65 CMV 0## 3 TRB CASS… HomoSa… NLVPMVATV pp65 CMV 0## 4 TRB CASS… HomoSa… NLVPMVATV pp65 CMV 0## 5 TRB CSAD… HomoSa… NLVPMVATV pp65 CMV 0## 6 TRB CASS… HomoSa… NLVPMVATV pp65 CMV 0## 7 TRB CASS… HomoSa… NLVPMVATV pp65 CMV 0## 8 TRB CASS… HomoSa… NLVPMVATV pp65 CMV 0## 9 TRB CSVE… HomoSa… NLVPMVATV pp65 CMV 0## 10 TRB CASS… HomoSa… NLVPMVATV pp65 CMV 0## # … with 4,486 more rows, and 12 more variables: v.segm <chr>, j.segm <chr>,## # v.end <dbl>, j.start <dbl>, mhc.a <chr>, mhc.b <chr>, mhc.class <chr>,## # reference.id <chr>, vdjdb.score <dbl>, Species <chr>, Chain <chr>,## # Pathology <chr>
# Check if everything is OK and there is no other epitopestable(vdjdb$antigen.epitope)#### NLVPMVATV## 4496
参考来源:https://immunarch.com/articles/web_only/v11_db.html

若有收获,就点个赞吧
0 人点赞
