Garnett包简介
Garnett是一个根据单细胞表达数据自动进行细胞类型分类注释的R包。Garnett使用单细胞表达数据和细胞类型定义(marker)文件,训练一个基于回归的分类器。一旦针对某一组织/样本类型训练成了一个分类器,它就可以将其应用于对其他相似组织的数据集进行分类注释。除了描述训练和分类的功能外,该网站还旨在成为一个存储以前训练出来的分类器仓库。
Garnett及其依赖包的安装
Garnett runs in the R statistical computing environment. You will need R version 3.5 or higher to install Garnett.
# Install from Github# Garnett builds upon a package called Monocle. Before installing Garnett, first install Monocle using Bioconductor:# First install Bioconductor and Monocleif (!requireNamespace("BiocManager"))install.packages("BiocManager")# 安装monocle包BiocManager::install(c("monocle"))# Next install a few more dependencies# 安装依赖包BiocManager::install(c('DelayedArray', 'DelayedMatrixStats', 'org.Hs.eg.db', 'org.Mm.eg.db'))# 使用devtools安装garnett包install.packages("devtools")devtools::install_github("cole-trapnell-lab/garnett")# Load Garnettlibrary(garnett)
Garnett包的工作流程
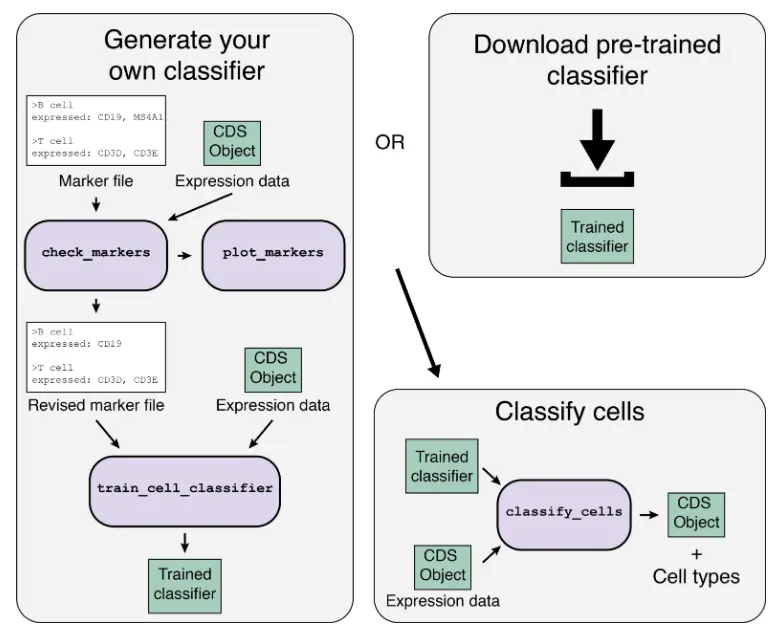
Garnett包的工作流程主要包括以下两个部分:
- Train/obtain the classifier: 我们可以下载现有的细胞分类器,或者训练自己的分类器。训练分类器时,Garnett首先解析一个标记文件(marker file),并选择一组训练细胞,然后训练一个多项分类器来区分不同的细胞类型。
- Classify cells: 接下来,Garnett将训练好的分类器应用于一组细胞,以生成细胞类型分配。Garnett还可以选择将分类扩展到类似的细胞,以生成一组独立的分群扩展类型分配。
1a. Using a pre-trained classifier 使用一个预先训练好的分类器
Garnett包工作的第一步是构建一个细胞类型分类器,我们既可以使用预先训练好的分类器,也可以单独训练一个自己的分类器。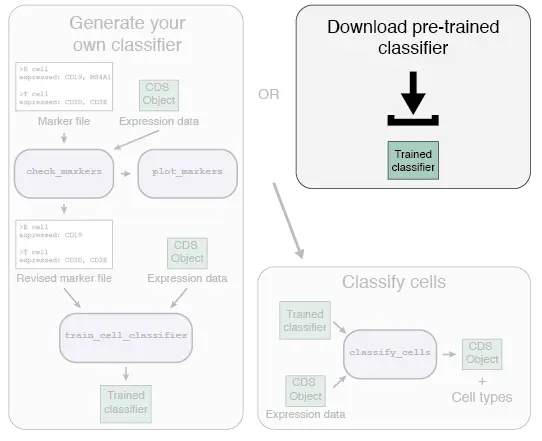
我们已经为各种生物和组织生成了一系列预训练好的分类器。如果您的数据类型存在预先训练好的分类器,我们建议您尝试一下,可以在此处找到可用分类器的列表(https://cole-trapnell-lab.github.io/garnett/classifiers/)。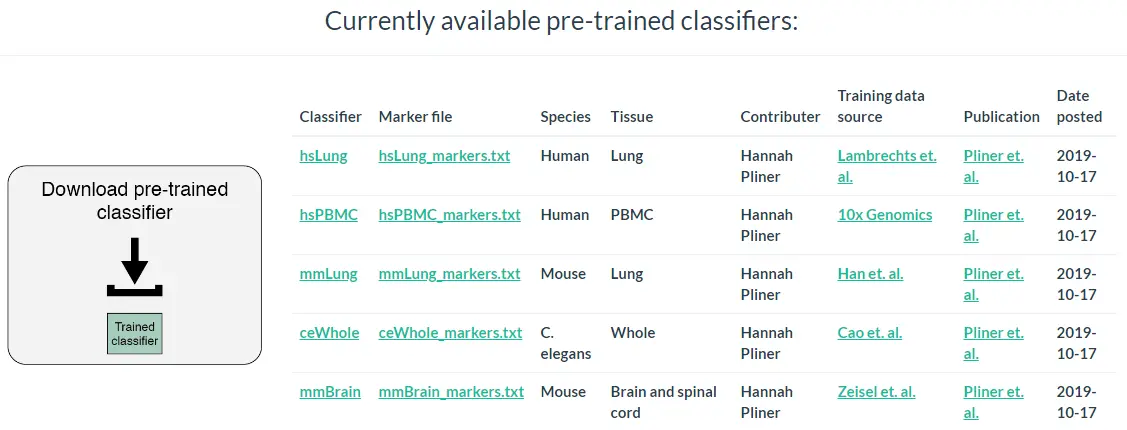
要使用预先训练好的分类器,请首先下载分类器,然后使用以下命令将其加载到您的R会话中:
classifier <- readRDS("path/to/classifier.RDS")
1b. Train your own classifier 训练一个自己的分类器
如果我们使用的组织类型不存在分类器,或者我们的数据中不包含预期的细胞类型,则需要训练生成一个自己的分类器。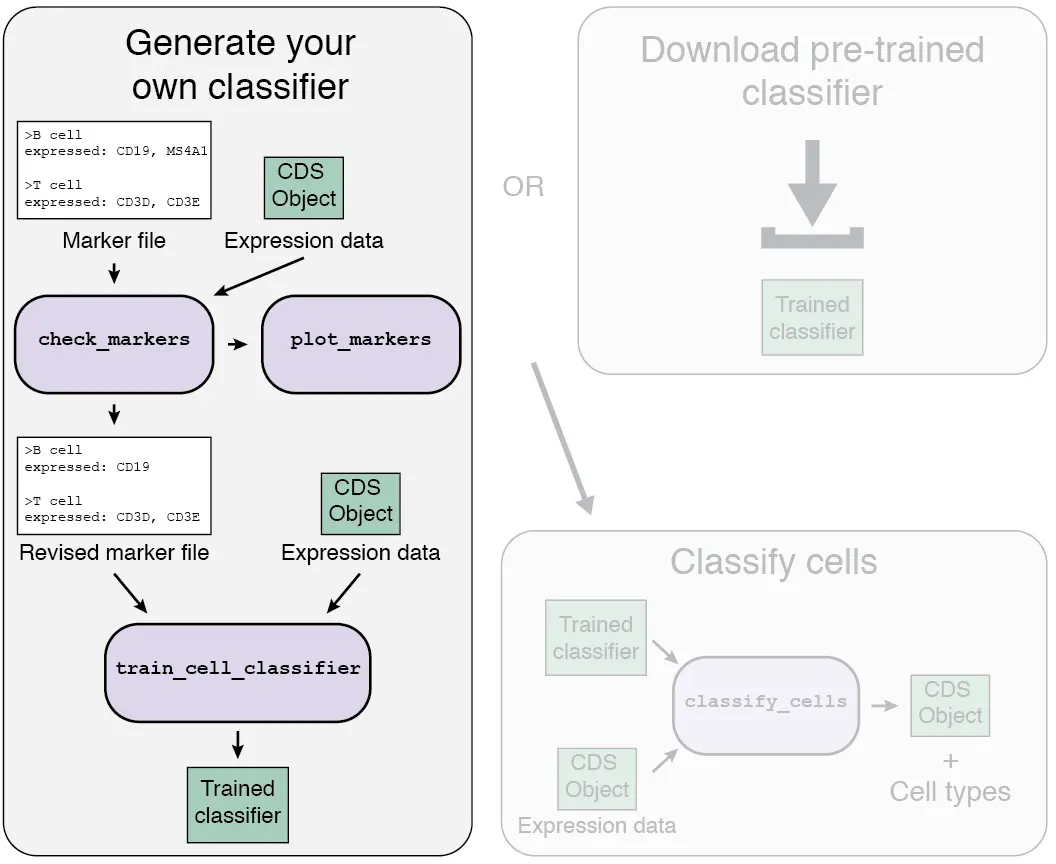
Loading your data
首先,我们加载单细胞的表达数据,并使用它训练一个细胞分类器。这里,我们使用10x平台测序的PBMC数据进行演示。
# load in the data
# NOTE: the 'system.file' file name is only necessary to read in included package data
library(monocle)
# 读取表达矩阵
mat <- Matrix::readMM(system.file("extdata", "exprs_sparse.mtx", package = "garnett"))
head(mat)
6 x 800 sparse Matrix of class "dgTMatrix"
[1,] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ......
[2,] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ......
[3,] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ......
[4,] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ......
[5,] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ......
[6,] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ......
.....suppressing 768 columns in show(); maybe adjust 'options(max.print= *, width = *)'
..............................
# 读取基因的metadata信息
fdata <- read.table(system.file("extdata", "fdata.txt", package = "garnett"))
head(fdata)
gene_short_name num_cells_expressed
MIR1302-10 MIR1302-10 0
FAM138A FAM138A 0
OR4F5 OR4F5 0
RP11-34P13.7 RP11-34P13.7 0
RP11-34P13.8 RP11-34P13.8 0
AL627309.1 AL627309.1 0
# 读取细胞的metadata信息
pdata <- read.table(system.file("extdata", "pdata.txt", package = "garnett"), sep="\t")
head(pdata)
tsne_1 tsne_2 Size_Factor FACS_type
AAGCACTGCACACA-1 3.840315 12.084191 0.5591814 B cells
GGCTCACTGGTCTA-1 9.970962 3.505393 0.5159340 B cells
AGCACTGATATCTC-1 3.459529 4.935273 0.6980284 B cells
ACACGTGATATTCC-1 1.743949 7.782671 0.8156310 B cells
ATATGCCTCTGCAA-1 5.783448 8.558898 1.1153280 B cells
TGACGAACCTATTC-1 10.792853 10.585274 0.6494699 B cells
row.names(mat) <- row.names(fdata)
colnames(mat) <- row.names(pdata)
# create a new CDS object
# 构建CDS对象
pd <- new("AnnotatedDataFrame", data = pdata)
fd <- new("AnnotatedDataFrame", data = fdata)
pbmc_cds <- newCellDataSet(as(mat, "dgCMatrix"),
phenoData = pd,
featureData = fd)
pbmc_cds
CellDataSet (storageMode: environment)
assayData: 32738 features, 800 samples
element names: exprs
protocolData: none
phenoData
sampleNames: AAGCACTGCACACA-1 GGCTCACTGGTCTA-1 ...
GTTGACGACCTTAT-1 (800 total)
varLabels: tsne_1 tsne_2 Size_Factor FACS_type
varMetadata: labelDescription
featureData
featureNames: MIR1302-10 FAM138A ... AC002321.1 (32738 total)
fvarLabels: gene_short_name num_cells_expressed
fvarMetadata: labelDescription
experimentData: use 'experimentData(object)'
Annotation:
# generate size factors for normalization later
pbmc_cds <- estimateSizeFactors(pbmc_cds)
Constructing a marker file
除了表达数据外,我们还需要另一个主要的输入文件(marker file)。标记文件中包含着细胞类型定义的列表,该文件告诉Garnett如何选择细胞来训练分类模型。每个细胞类型定义均以“>”号和细胞类型名称开头,后接一系列带有定义信息的行。定义行以关键字和“:”开头,不同条目之间用逗号分隔。
A simple valid example:
>B cells
expressed: CD19, MS4A1
>T cells
expressed: CD3D
通常,每个细胞类型定义可以包含三个主要部分,其中第一个部分是必须的。
Define the expression
细胞类型定义的第一个也是最重要的说明是其表达的marker基因。Garnett提供了几种用于指定marker基因的选项,详细信息如下。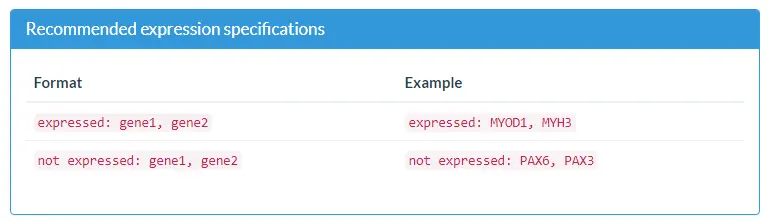
以上是为细胞类型指定marker基因的默认方法。使用此规范时,Garnett会计算每个细胞的marker基因的得分,并考虑到总体的表达水平和测序深度。

这是指定marker基因表达的另一种方法,如果我们知道了期望基因表达占据的确切范围,则该方法很有用。但是,一般而言,我们不建议使用这些规范,因为它们不会考虑每个细胞中的测序深度和整体的表达情况。
Define the meta data
除了表达信息,我们还可以使用元数据进一步细分细胞的类型定义。我们可以在此处指定数据中期望的任何子类型。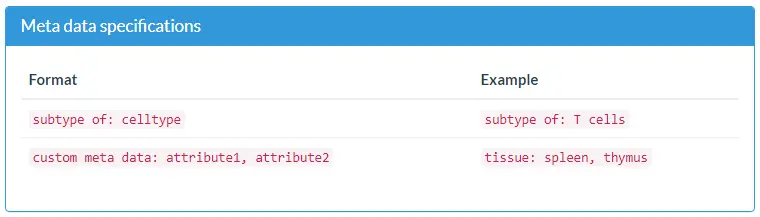
subtype of: 允许我们指定某一细胞类型是定义文件中另一种细胞类型的子类型。custom meta data: 允许我们为细胞类型定义提供任何其他的meta信息。
Provide your evidence
最后,我们强烈建议您记录如何选择的这些marker基因。为了便于跟踪,我们提供了一个附加的规范-reference:- 存储每种细胞类型的引文信息。添加一组URL或DOI,它们将包含在您的分类器中。
Add any comments
类似的,我们还提供了注释字符#号,因此我们还可以在细胞类型标记文件中添加一些注释信息。Garnett会忽略掉带有#号行之后的所有内容。
A more complex example:
>B cells
expressed: CD19, MS4A1
expressed above: CD79A 10
references: https://www.abcam.com/primary-antibodies/b-cells-basic-immunophenotyping, 10.3109/07420528.2013.775654
>T cells
expressed: CD3D
sample: blood # A meta data specification
>Helper T cells
expressed: CD4
subtype of: T cells
references: https://www.ncbi.nlm.nih.gov/pubmed/?term=12000723
Checking your markers
由于定义标记文件通常是该过程中最困难的部分,因此Garnett包提供了用于检查标记是否能正常工作的函数。相关的两个函数是check_markers和plot_markers。check_markers函数将生成有关标记的信息表,plot_markers函数绘制出最相关的信息。
>B cells
expressed: CD19, MS4A1, CD79A, ACTN, ACTB
references: https://www.ncbi.nlm.nih.gov/pubmed/?term=23688120,
https://www.ncbi.nlm.nih.gov/pubmed/?term=21149806
>T cells
expressed: CD3D, CD3E, CD3G, PTPRC
references: https://www.ncbi.nlm.nih.gov/pubmed/?term=1534551
>CD4 T cells
expressed: CD4, FOXP3, IL2RA, IL7R
subtype of: T cells
>CD8 T cells
expressed: CD8A, CD8B
subtype of: T cells

library(org.Hs.eg.db)
marker_file_path <- system.file("extdata", "pbmc_bad_markers.txt",
package = "garnett")
# 使用check_markers函数检查标记基因的信息
marker_check <- check_markers(pbmc_cds, marker_file_path,
db=org.Hs.eg.db,
cds_gene_id_type = "SYMBOL",
marker_file_gene_id_type = "SYMBOL")
head(marker_check)
marker_gene gene_id parent cell_type in_cds nominates
1 CD3D ENSG00000167286 root T cells TRUE 269
2 CD3E ENSG00000198851 root T cells TRUE 295
3 CD3G ENSG00000160654 root T cells TRUE 104
4 PTPRC ENSG00000081237 root T cells TRUE 179
5 CD4 ENSG00000010610 root T cells TRUE 25
6 FOXP3 ENSG00000049768 root T cells TRUE 8
total_nominated exclusion_dismisses inclusion_ambiguates most_overlap
1 486 14 10 B cells
2 486 24 16 B cells
3 486 1 1 B cells
4 486 97 94 B cells
5 486 0 0 <NA>
6 486 0 0 <NA>
ambiguity marker_score summary
1 0.037174721 11.7329351 Ok
2 0.054237288 9.4492763 Ok
3 0.009615385 10.9093843 Ok
4 0.525139665 0.6882554 High ambiguity?
5 0.000000000 5.1440329 Ok
6 0.000000000 1.6460905 Low nomination?
# 使用plot_markers函数对marker基因的信息进行可视化
plot_markers(marker_check)
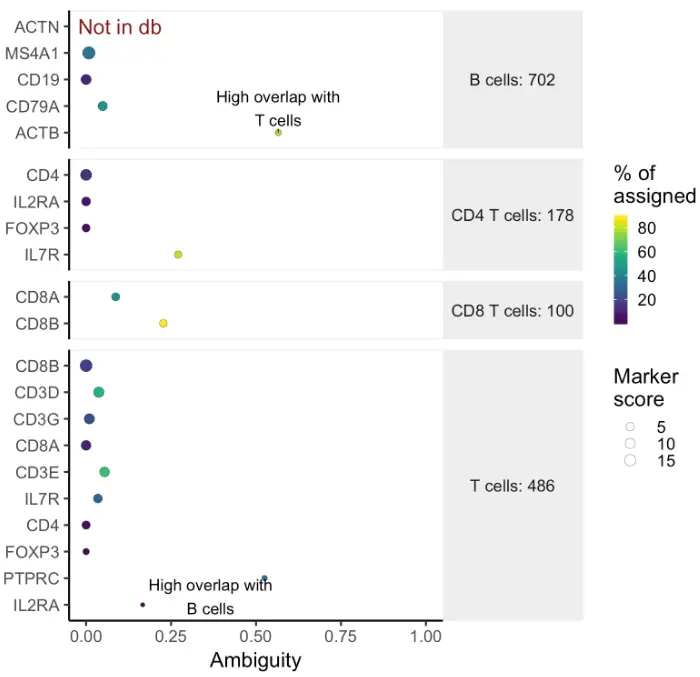
这个标记图中提供了一些关于所选marker基因是否正确的关键信息。首先,红色标记的“not in db”告诉我们ACTN marker基因在org.Hs.eg.db注释中没有提供“SYMBOL”信息。接下来,x轴显示了每个marker基因的模糊度评分(ambiguity score)。在本例中,ACTB和PTPRC marker基因具有较高的模糊度得分,应将其排除。
注意:check_markers函数输出的值和plot_markers函数绘制的值是分类器选择的细胞数量的估计值。然而,它使用启发式快速找到候选细胞,并不能完全匹配标记所选择的细胞。请使用这些数字作为相对的度量,而不是训练集的绝对表示。
#pbmc_test.txt
>B cells
expressed: CD19, MS4A1, CD79A
references: https://www.ncbi.nlm.nih.gov/pubmed/?term=23688120, https://www.ncbi.nlm.nih.gov/pubmed/?term=21149806
>T cells
expressed: CD3D, CD3E, CD3G
references: https://www.ncbi.nlm.nih.gov/pubmed/?term=1534551
>CD4 T cells
expressed: CD4, FOXP3, IL2RA, IL7R
subtype of: T cells
>CD8 T cells
expressed: CD8A, CD8B
subtype of: T cells
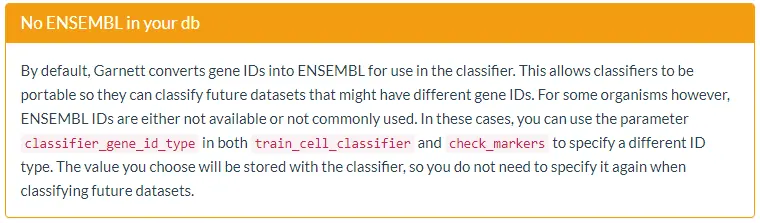
Train the classifier
构建好细胞类型marker标记文件后,我们就可以使用train_cell_classifier函数训练分类器了,其参数与check_markers函数的参数类似。下面,我们更改了默认的一个num_unknown参数。 该参数告诉Garnett应该与多少个outgroup细胞进行比较,默认值为500,但是在此数据集中细胞数量较少,因此也设置的小一些。
library(org.Hs.eg.db)
set.seed(260)
marker_file_path <- system.file("extdata", "pbmc_test.txt",
package = "garnett")
# 使用train_cell_classifier函数进行训练构建分类器
pbmc_classifier <- train_cell_classifier(cds = pbmc_cds,
marker_file = marker_file_path,
db=org.Hs.eg.db,
cds_gene_id_type = "SYMBOL",
num_unknown = 50,
marker_file_gene_id_type = "SYMBOL")
There are 4 cell type definitions
training_sample
B cells T cells Unknown
200 200 50
Model training finished.
training_sample
CD4 T cells CD8 T cells Unknown
72 44 50
Model training finished.
# 查看训练的分类结果
pbmc_classifier
An object of class "garnett_classifier"
Slot "classification_tree":
IGRAPH 74f63cf DN-- 5 4 --
+ attr: name (v/c), classify_func (v/x), model (v/x)
+ edges from 74f63cf (vertex names):
[1] root ->B cells root ->T cells T cells->CD4 T cells
[4] T cells->CD8 T cells
Slot "cell_totals":
[1] 2.615075
Slot "gene_id_type":
[1] "ENSEMBL"
Slot "references":
$`B cells`
[1] "https://www.ncbi.nlm.nih.gov/pubmed/?term=23688120"
[2] "https://www.ncbi.nlm.nih.gov/pubmed/?term=21149806"
$`T cells`
[1] "https://www.ncbi.nlm.nih.gov/pubmed/?term=1534551"
head(pData(pbmc_cds))
# tsne_1 tsne_2 Size_Factor FACS_type
# AAGCACTGCACACA-1 3.840315 12.084191 0.5591814 B cells
# GGCTCACTGGTCTA-1 9.970962 3.505393 0.5159340 B cells
# AGCACTGATATCTC-1 3.459529 4.935273 0.6980284 B cells
# ACACGTGATATTCC-1 1.743949 7.782671 0.8156310 B cells
# ATATGCCTCTGCAA-1 5.783448 8.558898 1.1153280 B cells
# TGACGAACCTATTC-1 10.792853 10.585274 0.6494699 B cells
运行完train_cell_classifier函数后,输出对象中的“garnett_classifier”类型将包含对细胞进行分类所需的所有信息。
Viewing the classification genes
Garnett使用多项式弹性网络回归(multinomial elastic-net regression)训练进行细胞类型分类。这意味着选择某些基因作为区分细胞类型的相关基因。选择哪些基因可能会令人感兴趣,因此Garnett提供了访问所选基因的功能。
Garnett使用get_feature_genes函数查看所选的相关基因,参数是分类器,想查看哪个节点(node)(如果分类树是分层的)—使用“root”作为顶部节点,其他节点使用父细胞类型名称,使用db参数指定参考物种。如果设置convert_ids = TRUE,则该函数将自动将基因id转换为SYMBOL。
feature_genes <- get_feature_genes(pbmc_classifier,
node = "root",
db = org.Hs.eg.db)
head(feature_genes)
# B cells T cells Unknown
#(Intercept) -6.51829349 2.6403157858 3.877977704
#CD74 0.04660324 -0.0529185035 0.006315261
#MS4A1 2.42667982 -2.2194139450 -0.207265871
#CD19 4.33990156 -2.4210500042 -1.918851560
#CD79A 1.09667783 -0.7008461805 -0.395831654
#IGLL5 -0.00103241 -0.0001045559 0.001136966
Viewing references
上面,我们解释了如何在标记文件中包含关于如何选择标记的文档。为了获取这些信息—查看如何为已经训练好的分类器选择标记—可以使用get_classifier_references函数。除了分类器之外,还有一个额外的cell_type可选参数。如果传递细胞类型的名称,则只会打印该细胞类型的引用,否则将全部打印。
get_classifier_references(pbmc_classifier)
#$`B cells`
#[1] "https://www.ncbi.nlm.nih.gov/pubmed/?term=23688120" "https://www.ncbi.nlm.nih.gov/pubmed/?term=21149806"
#$`T cells`
#[1] "https://www.ncbi.nlm.nih.gov/pubmed/?term=1534551"
Submitting a classifier
我们鼓励您向我们提交高质量的分类器,以便我们可以将其提供给社区。为此,请打开一个特刊并在Garnett github存储库中填写表单。单击此处(https://github.com/cole-trapnell-lab/garnett/issues),然后单击“New issue”按钮开始使用!
参考来源:https://cole-trapnell-lab.github.io/garnett/docs/#1b-train-your-own-classifier

若有收获,就点个赞吧
0 人点赞
