本次教程学习使用
scater包进行单细胞转录组数据降维与常用的一些可视化方法。
- plotExpression: plot cell expression levels for one or more genes;
- plotReducedDim: plot (and/or calculate) reduced dimension coordinates;
加载所需的R包和示例数据
library(scater)data("sc_example_counts")data("sc_example_cell_info")# 构建SingleCellExperiment对象example_sce <- SingleCellExperiment(assays = list(counts = sc_example_counts),colData = sc_example_cell_info)# 进行数据归一化example_sce <- normalize(example_sce)example_sce## class: SingleCellExperiment## dim: 2000 40## metadata(1): log.exprs.offset## assays(2): counts logcounts## rownames(2000): Gene_0001 Gene_0002 ... Gene_1999 Gene_2000## rowData names(0):## colnames(40): Cell_001 Cell_002 ... Cell_039 Cell_040## colData names(4): Cell Mutation_Status Cell_Cycle Treatment## reducedDimNames(0):## spikeNames(0):
基因表达量的可视化
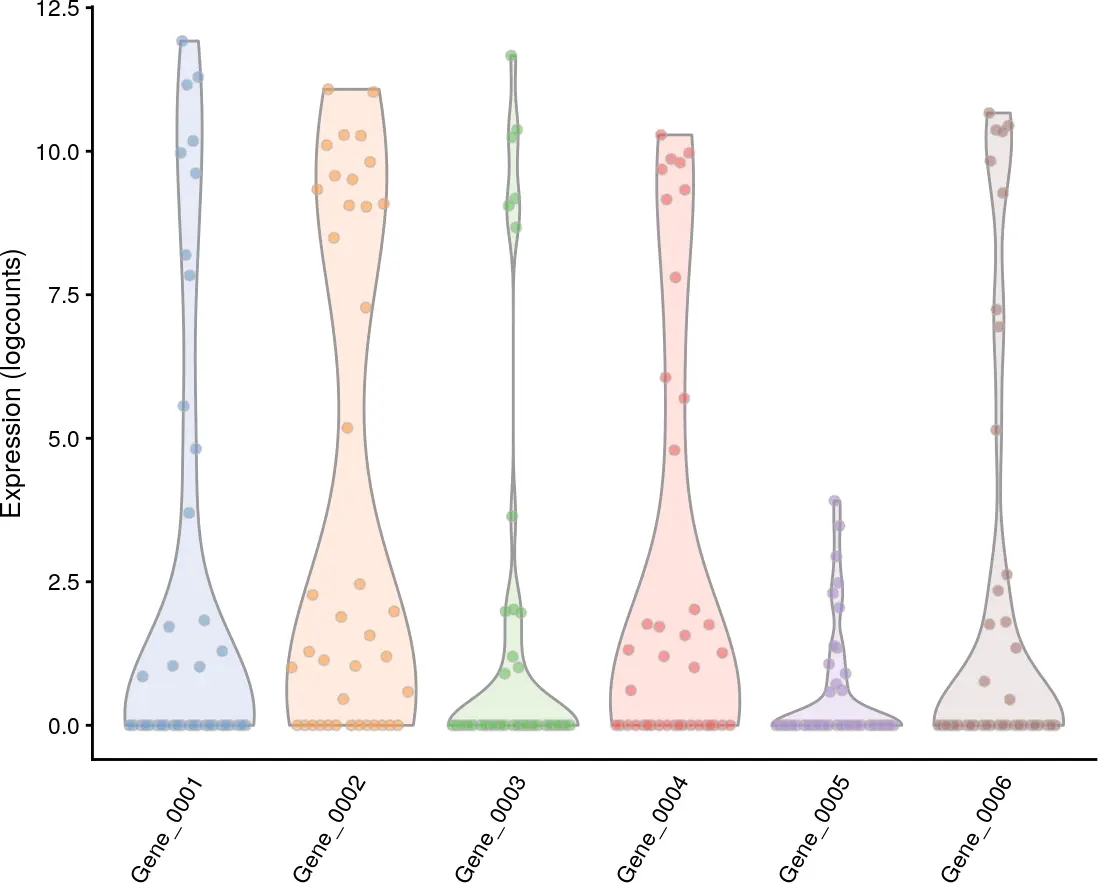
在scater包中,我们使用plotExpression函数对(特征)基因的表达量进行可视化展示。默认情况下,它使用归一化后的“logcounts”值进行可视化,我们也可以通过exprs_values参数对其进行更改。
plotExpression(example_sce, rownames(example_sce)[1:6],x = "Mutation_Status", exprs_values = "logcounts")
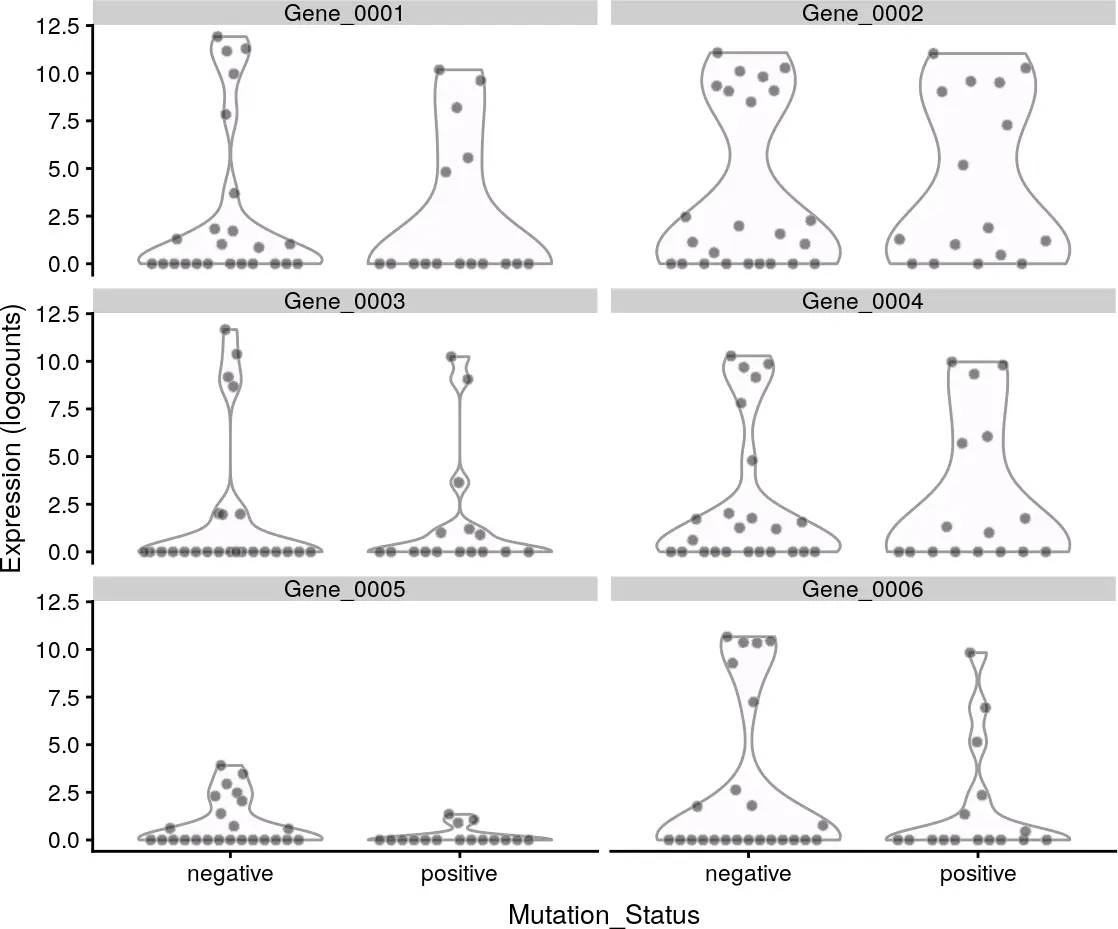
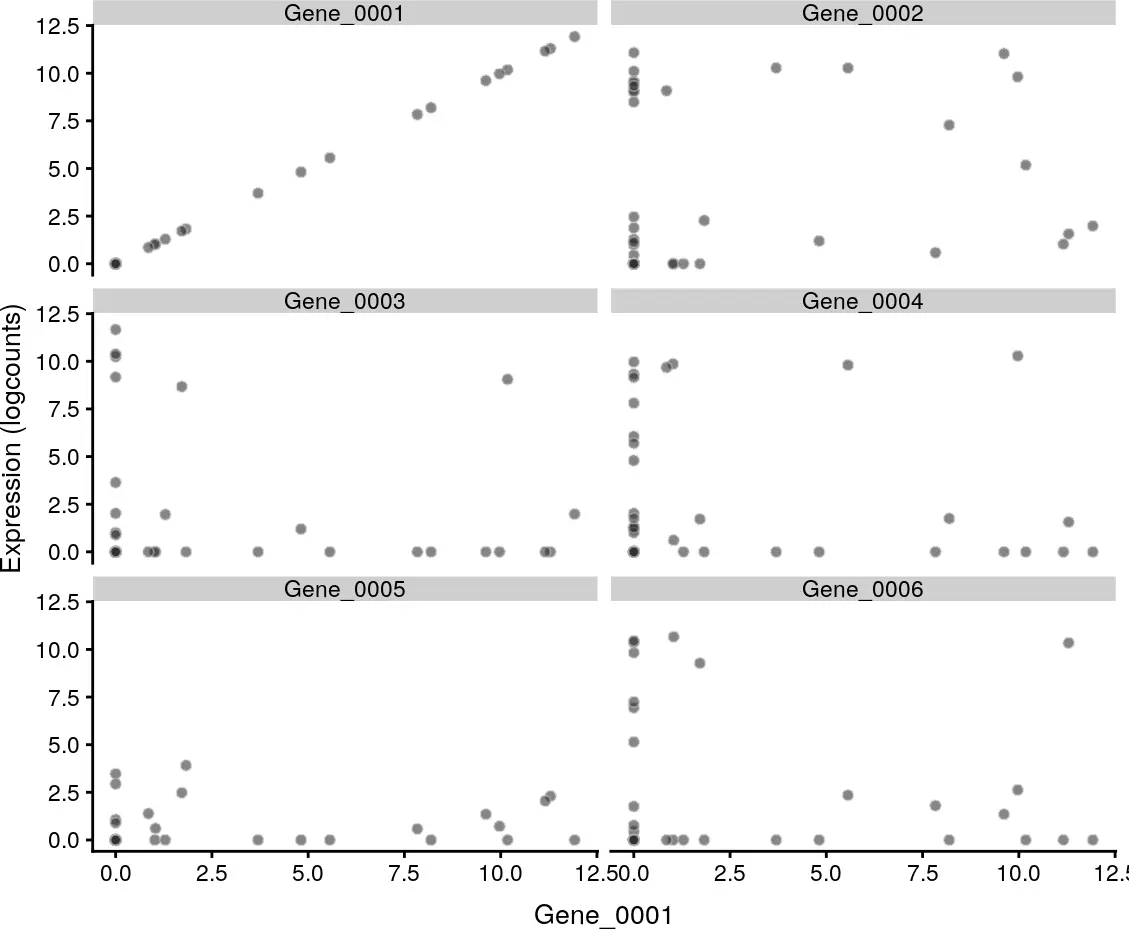
设置x参数确定在x轴上显示的协变量,它可以是列元数据中的字段,也可以是某个基因的名称。对于分类协变量,将产生如上图所示的分组小提琴图;而对于连续协变量,将在每个面板中生成散点图,如下图所示。
plotExpression(example_sce, rownames(example_sce)[1:6],x = "Gene_0001")
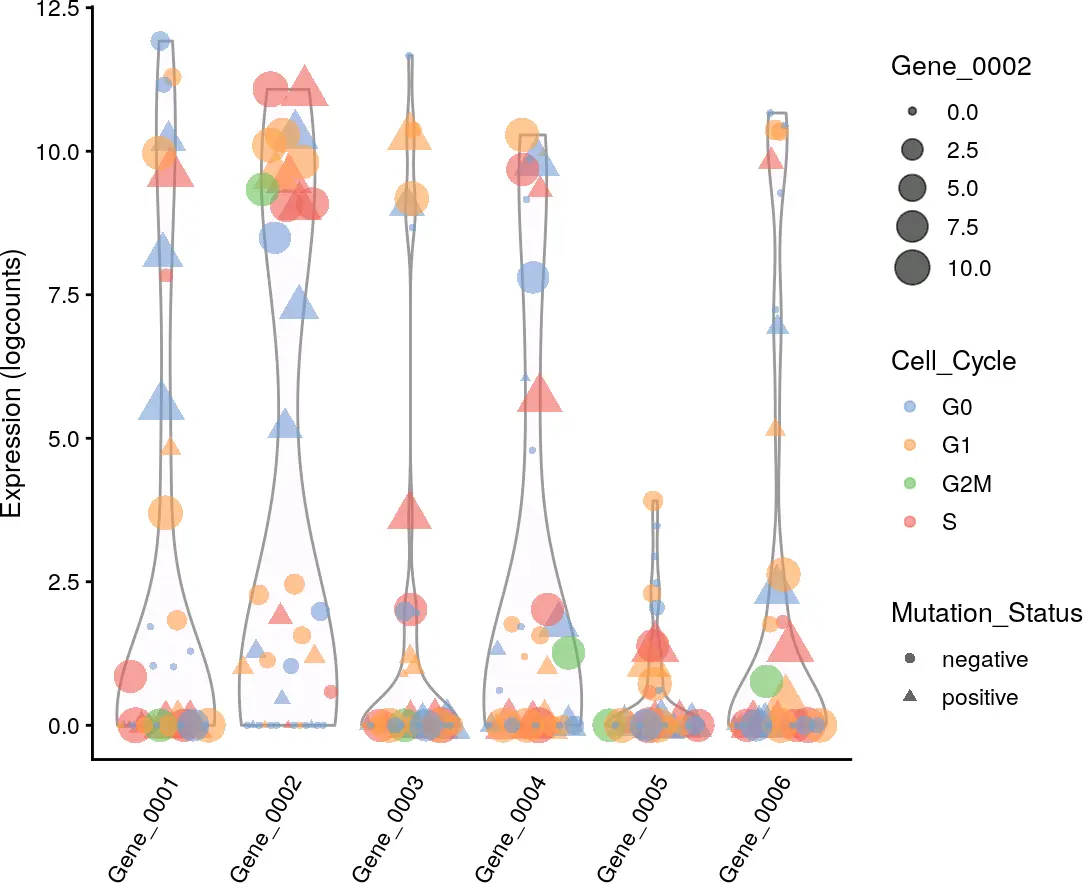
当然,我们也可以通过colour_by,shape_by和size_by等参数调整点的颜色,形状和大小。
plotExpression(example_sce, rownames(example_sce)[1:6],
colour_by = "Cell_Cycle", shape_by = "Mutation_Status",
size_by = "Gene_0002")
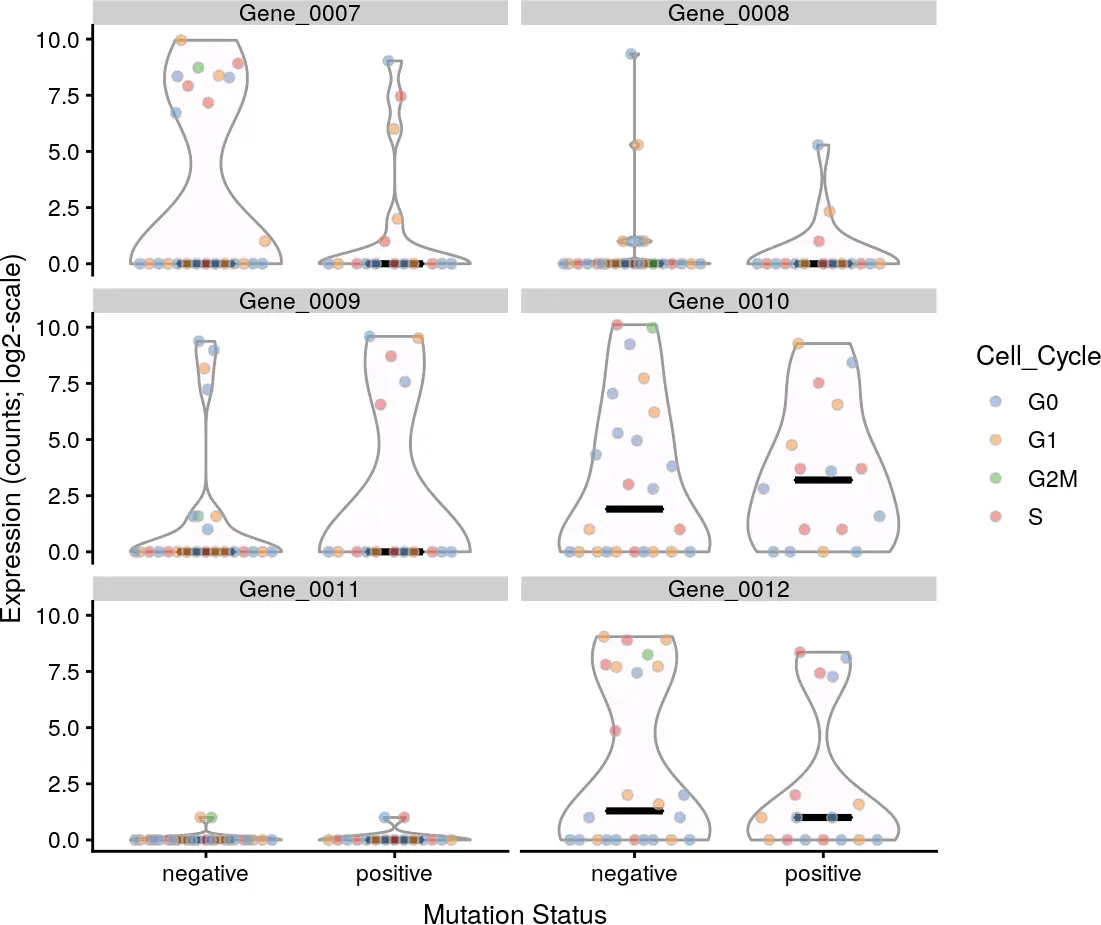
对于分类协变量x,我们还可以通过设置show_median = TRUE参数,在小提琴图上显示每组表达水平的中位数。
plotExpression(example_sce, rownames(example_sce)[7:12],
x = "Mutation_Status", exprs_values = "counts",
colour = "Cell_Cycle", show_median = TRUE,
xlab = "Mutation Status", log = TRUE)
如果不设置x参数,plotExpression函数将直接绘制一组所选基因的表达量的小提琴图。
plotExpression(example_sce, rownames(example_sce)[1:6])
数据的降维与可视化
scater包可以使用多种方法(PCA, tSNE, UMAP和diffussion maps)对单细胞转录组数据进行降维处理,降维后的结果存储在reducedDims slot中,可以使用plotReducedDim函数对降维后的结果进行可视化展示,并通过use_dimred参数设置降维的方法。
使用PCA方法进行数据降维可视化
scater包使用runPCA函数进行PCA降维处理,通过plotPCA函数进行降维可视化展示,也可以使用plotReducedDim函数对PCA降维后的结果进行可视化展示。
# 使用runPCA函数进行PCA降维处理
example_sce <- runPCA(example_sce)
reducedDimNames(example_sce)
## [1] "PCA"
# 使用plotReducedDim函数对降维结果进行可视化展示,use_dimred参数设置降维的方法
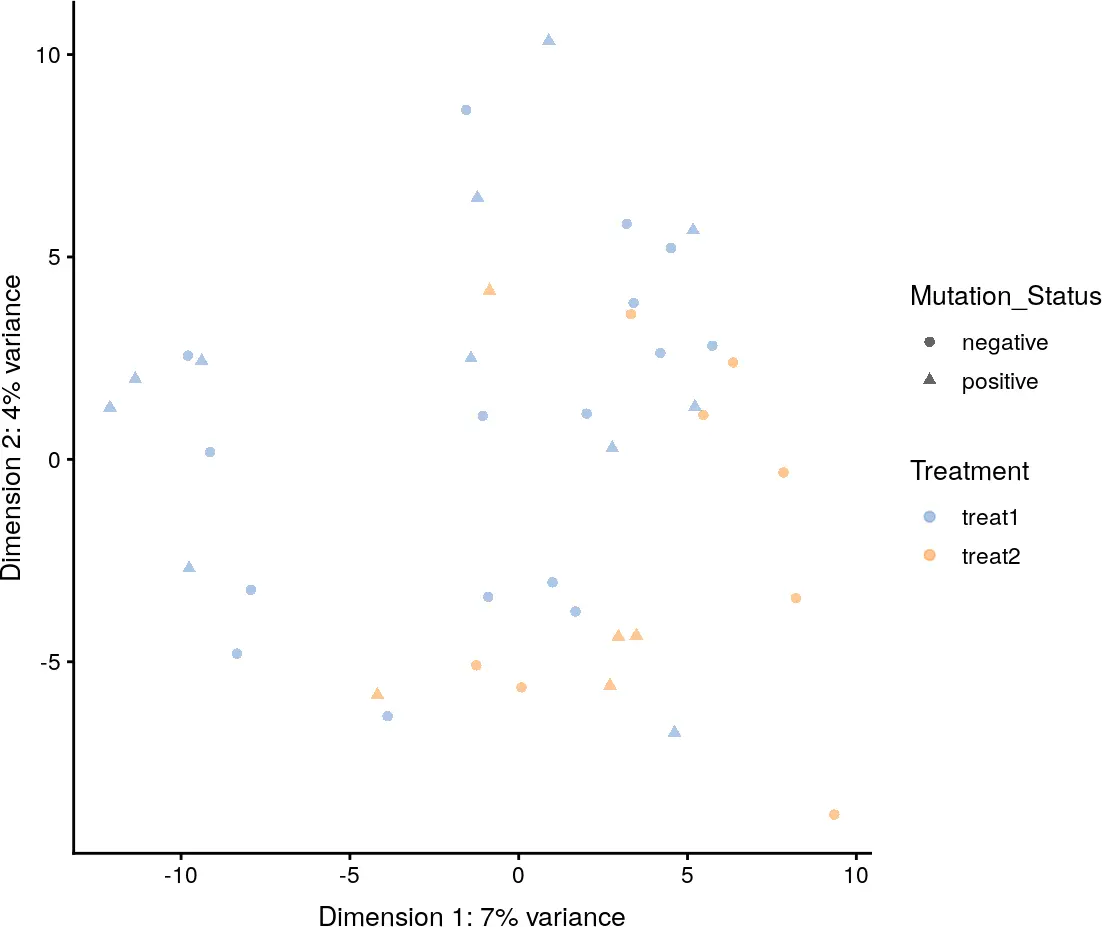
plotReducedDim(example_sce, use_dimred = "PCA",
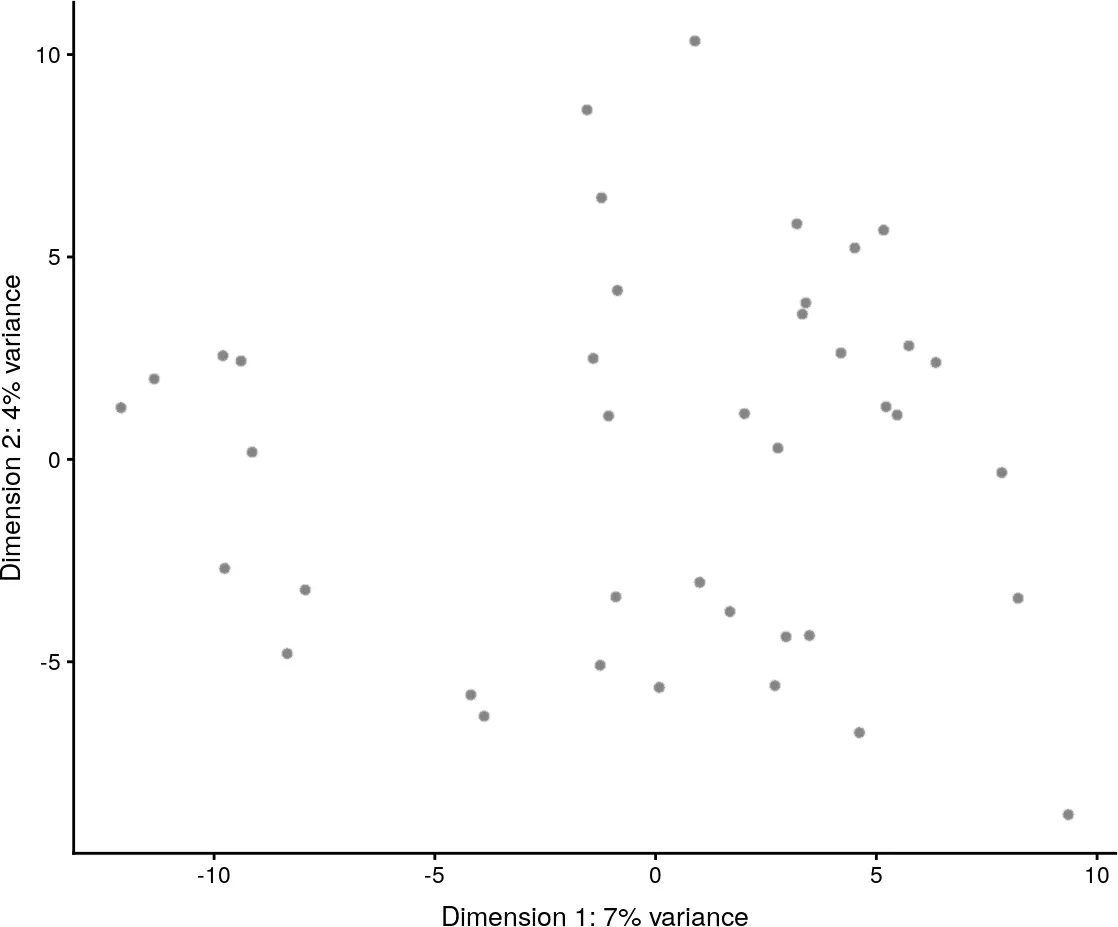
colour_by = "Treatment", shape_by = "Mutation_Status")
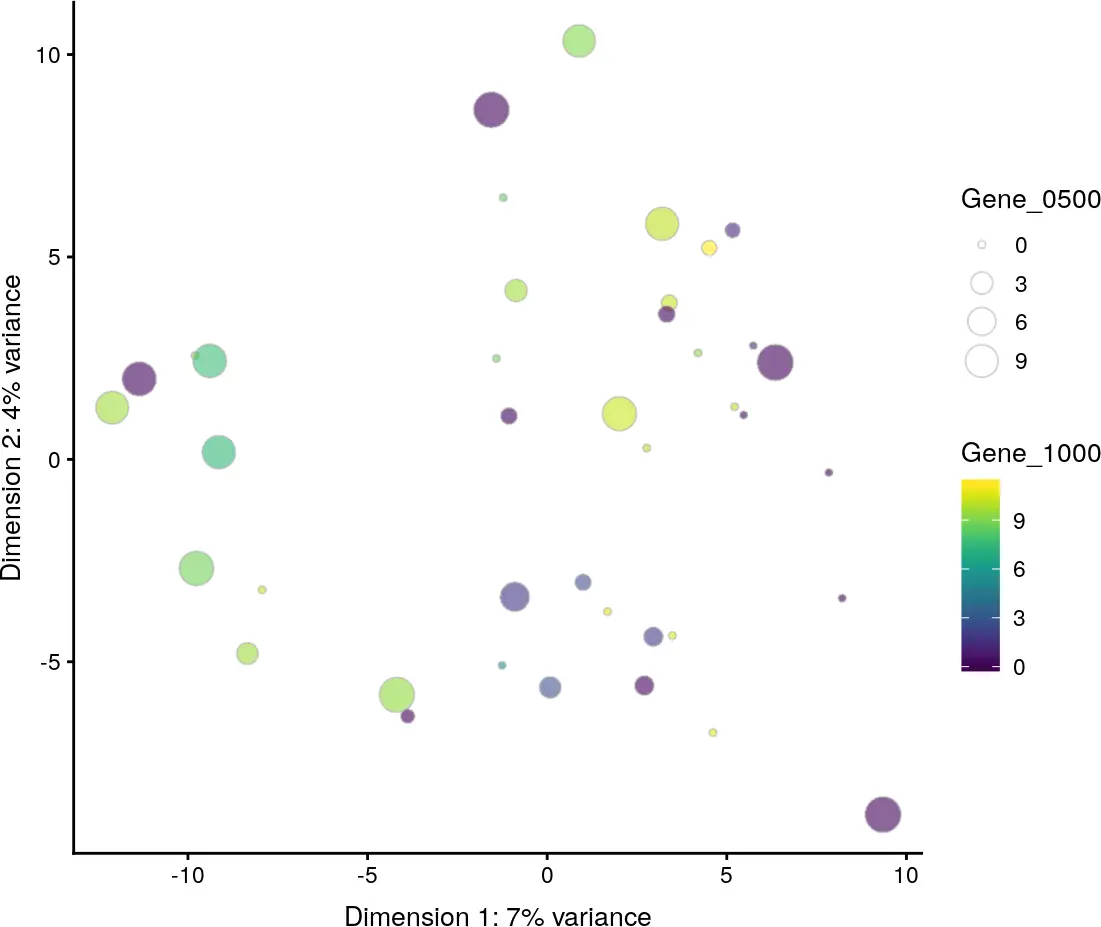
plotReducedDim(example_sce, use_dimred = "PCA",
colour_by = "Gene_1000", size_by = "Gene_0500")
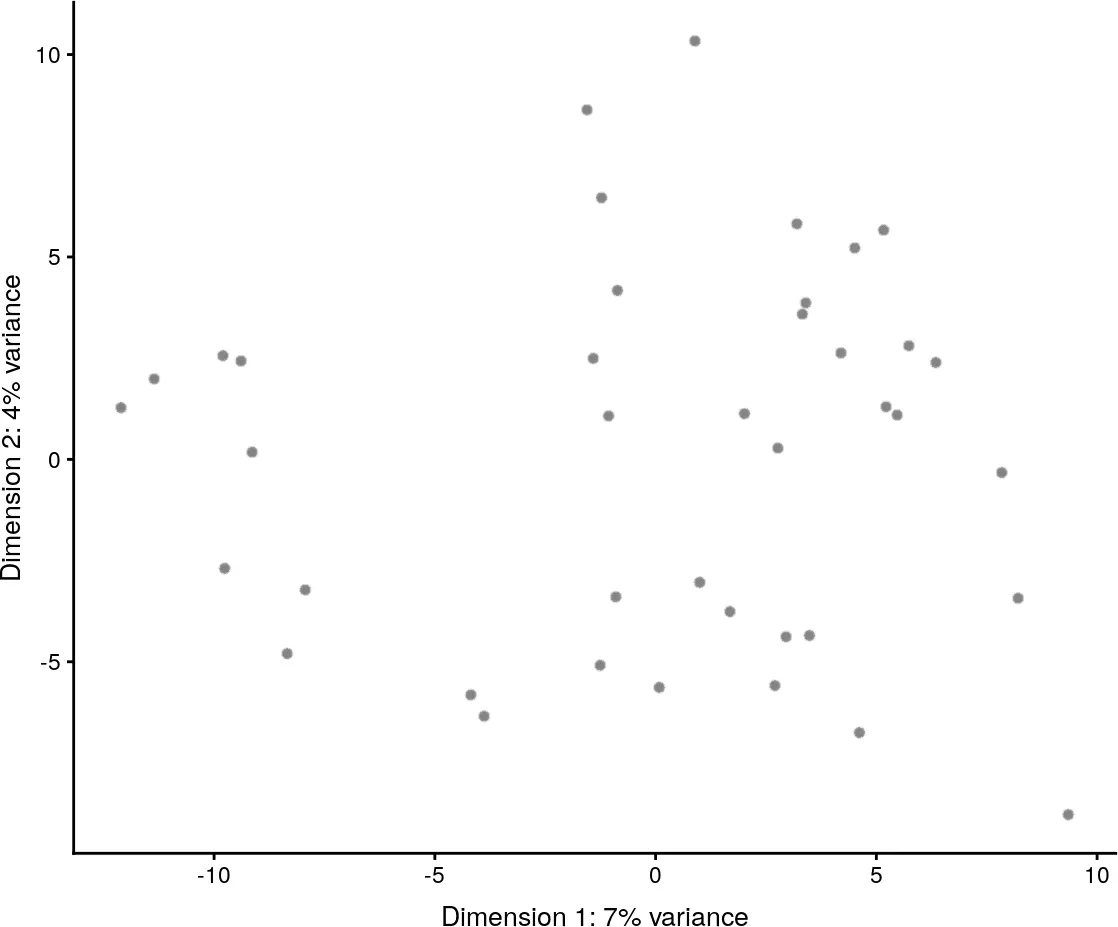
使用plotPCA函数对PCA降维的结果进行可视化展示,默认会对reduceDims slot中的PCA降维结果的前两个主成分进行可视化。
plotPCA(example_sce)
如果在使用plotPCA函数时不存在预先计算好的“PCA”结果,该函数将会自动调用runPCA函数计算PCA降维的结果。
默认情况下,runPCA函数会使用所有细胞中最高变化的500个基因的表达量的log-counts值来执行PCA降维处理,也可以通过ntop参数设置使用的高可变基因的数量。或者,通过feature_set参数设置用于PCA降维处理的一组特定基因。
example_sce2 <- runPCA(example_sce,
feature_set = rowData(example_sce)$is_feature_control)
plotPCA(example_sce2)
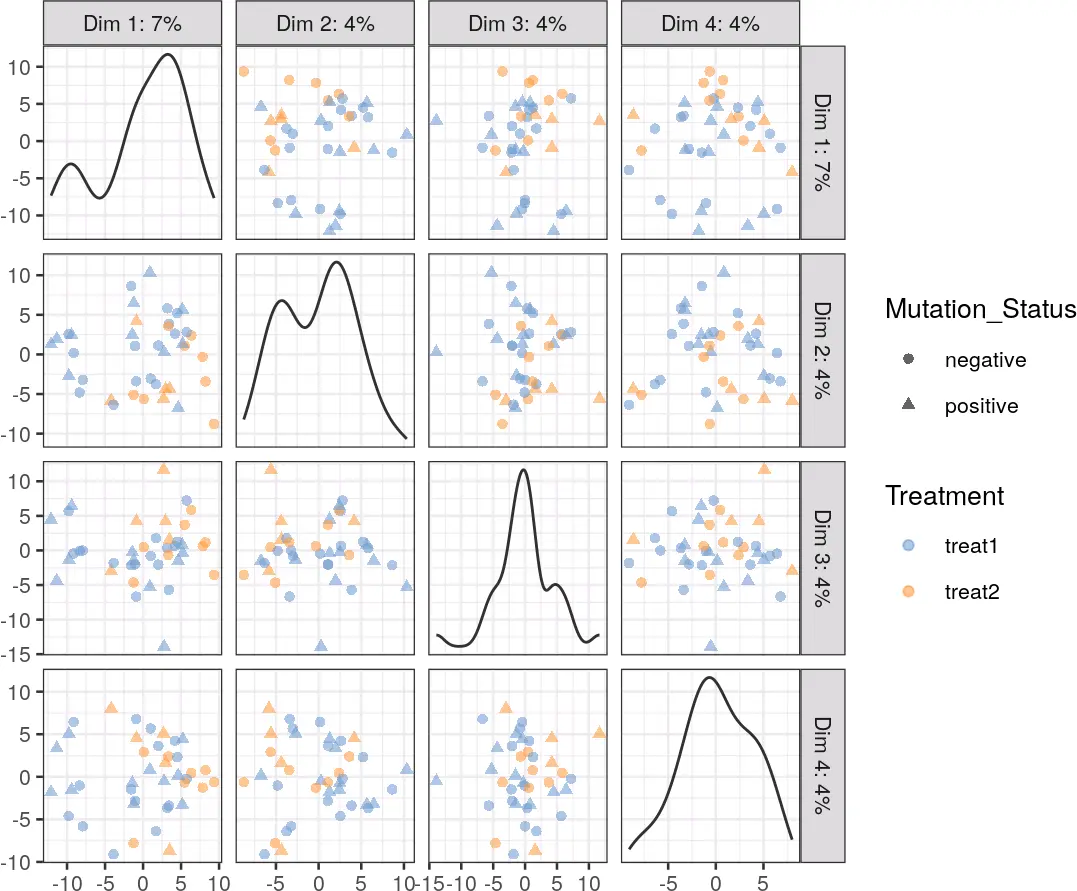
Multiple components can be plotted in a series of pairwise plots. When more than two components are plotted, the diagonal boxes in the scatter plot matrix show the density for each component.
example_sce <- runPCA(example_sce, ncomponents=20)
plotPCA(example_sce, ncomponents = 4, colour_by = "Treatment",
shape_by = "Mutation_Status")
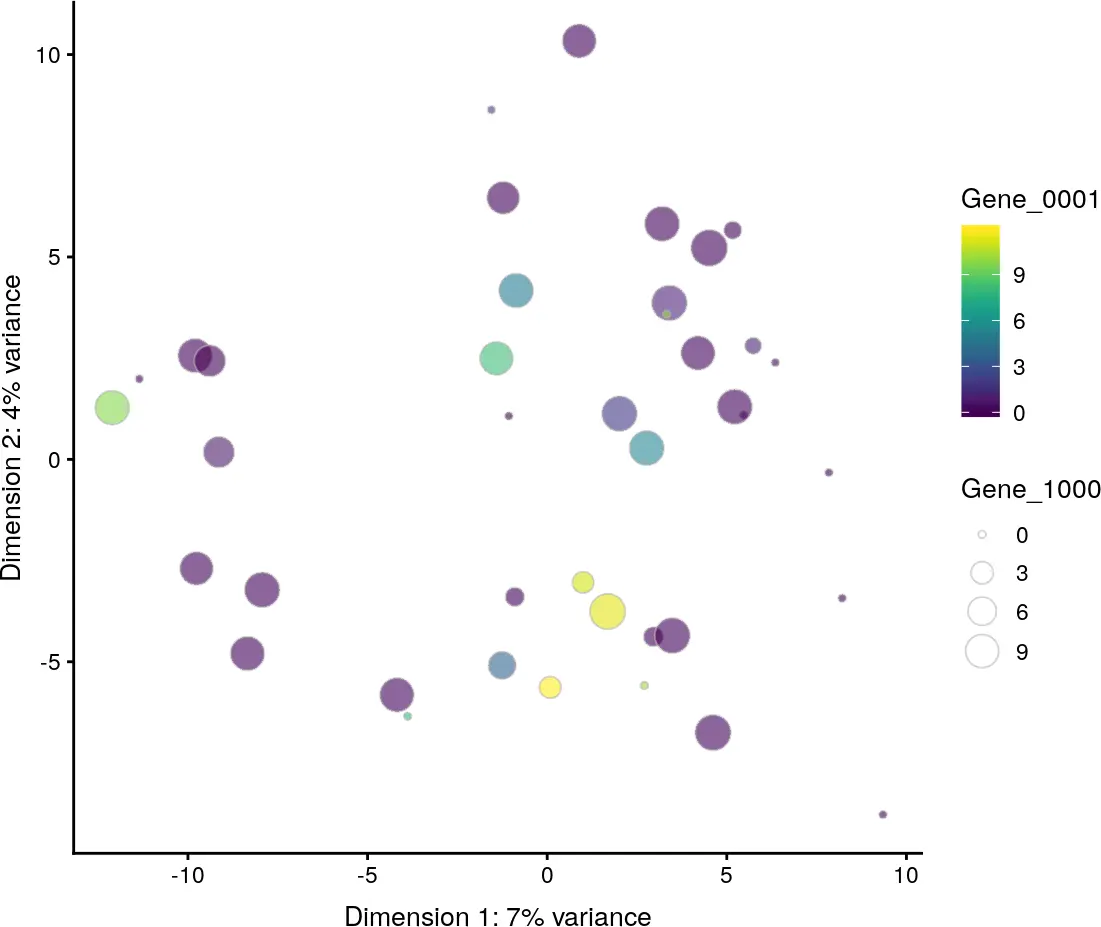
plotPCA(example_sce, colour_by = "Gene_0001", size_by = "Gene_1000")
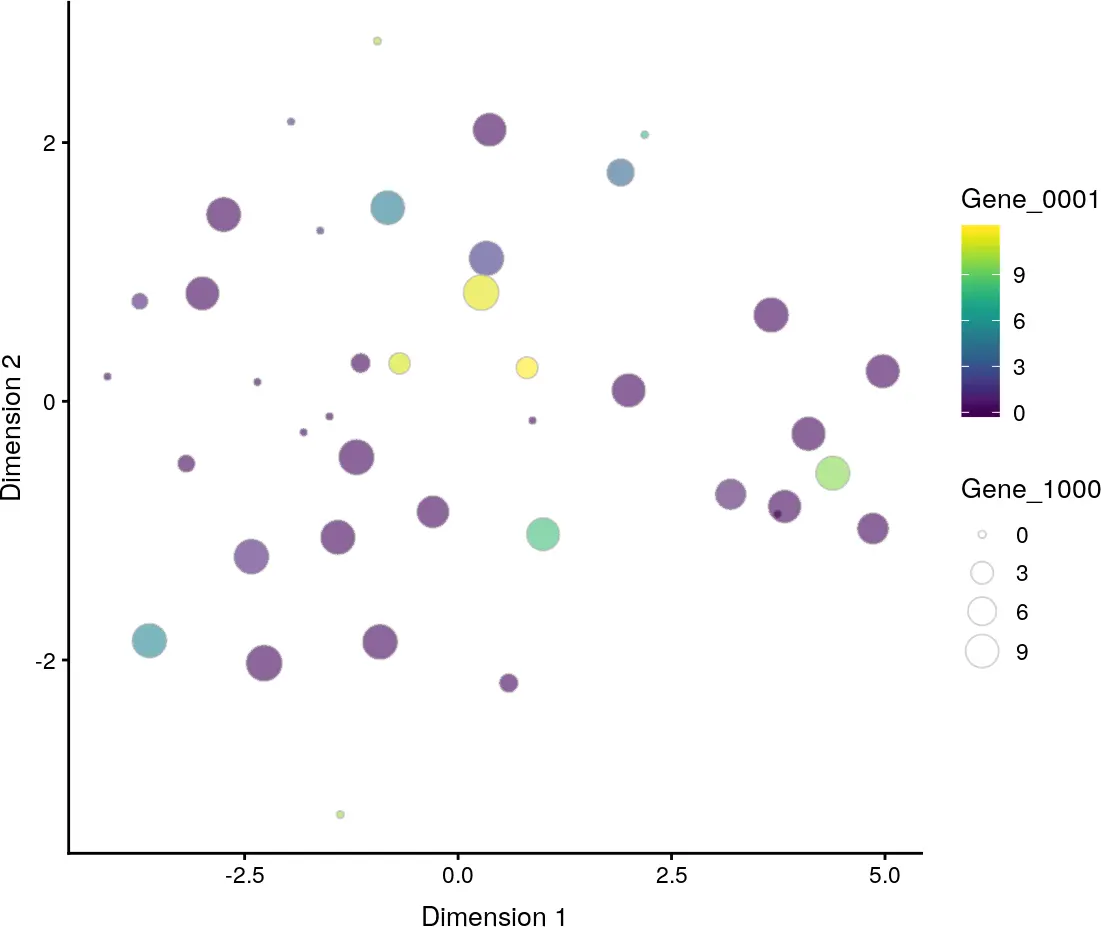
使用tSNE方法进行数据降维可视化
t-SNE方法被广泛用于复杂的单细胞数据集的降维可视化处理,scater通过Rtsne包使用runTSNE函数进行降维处理,获得tSNE降维后的坐标信息,使用plotTSNE函数可视化tSNE降维后的结果。
# Perplexity of 10 just chosen here arbitrarily.
set.seed(1000)
# 使用runTSNE函数进行tSNE数据降维处理
example_sce <- runTSNE(example_sce, perplexity=10)
# 使用plotTSNE函数将降维可视化展示
plotTSNE(example_sce, colour_by = "Gene_0001", size_by = "Gene_1000")
当然,我们也可以使用预先计算好的PCA降维结果作为t-SNE算法的输入,这可以使用low-rank approximation的表达矩阵提升计算的速度,也可以降低随机噪音。
set.seed(1000)
example_sce <- runTSNE(example_sce, perplexity=10, use_dimred="PCA", n_dimred=10)
plotTSNE(example_sce, colour_by="Treatment")
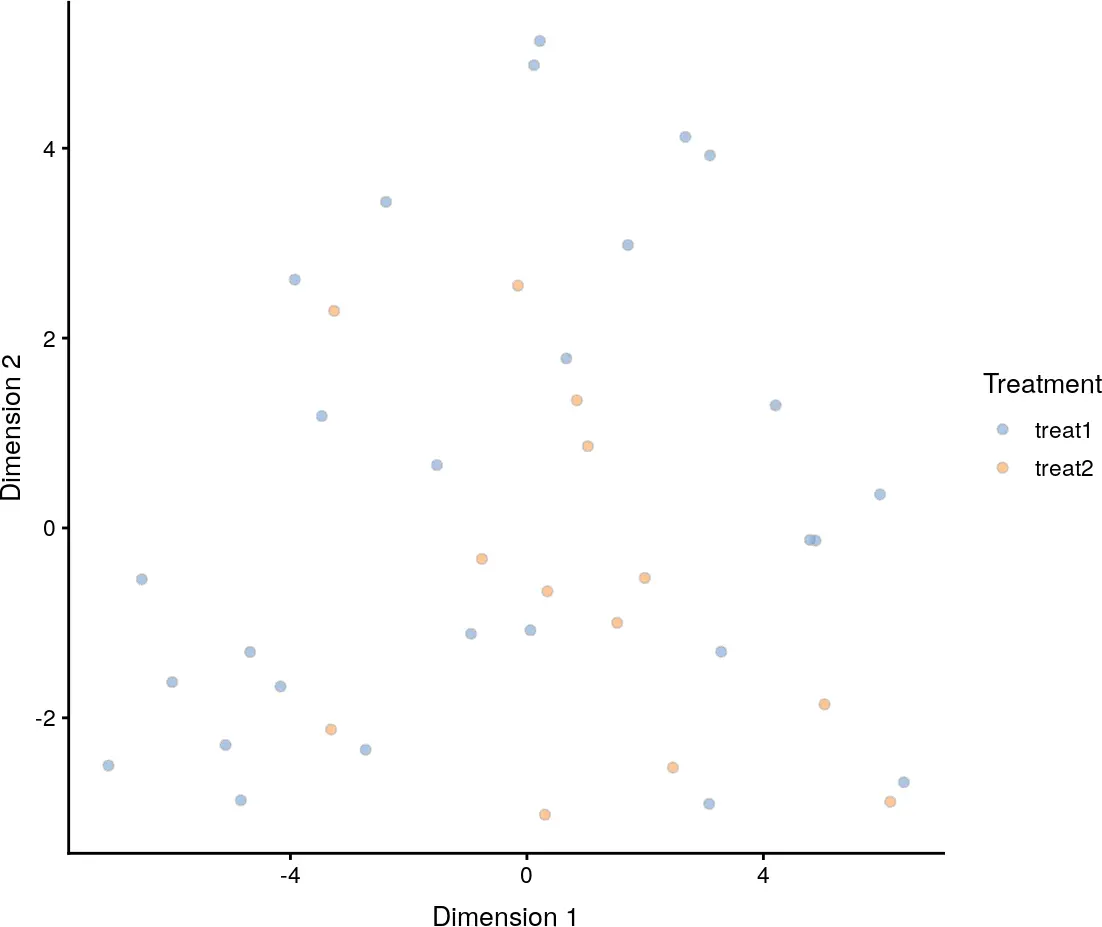
使用diffusion maps方法进行数据降维可视化
scater通过density包使用runDiffusionMap函数进行diffusion maps降维处理,并使用plotDiffusionMap函数对降维后的结果进行可视化展示。
# 使用runDiffusionMap函数进行降维处理
example_sce <- runDiffusionMap(example_sce)
使用plotDiffusionMap函数对降维结果进行可视化展示
plotDiffusionMap(example_sce, colour_by = "Gene_0001", size_by = "Gene_1000")
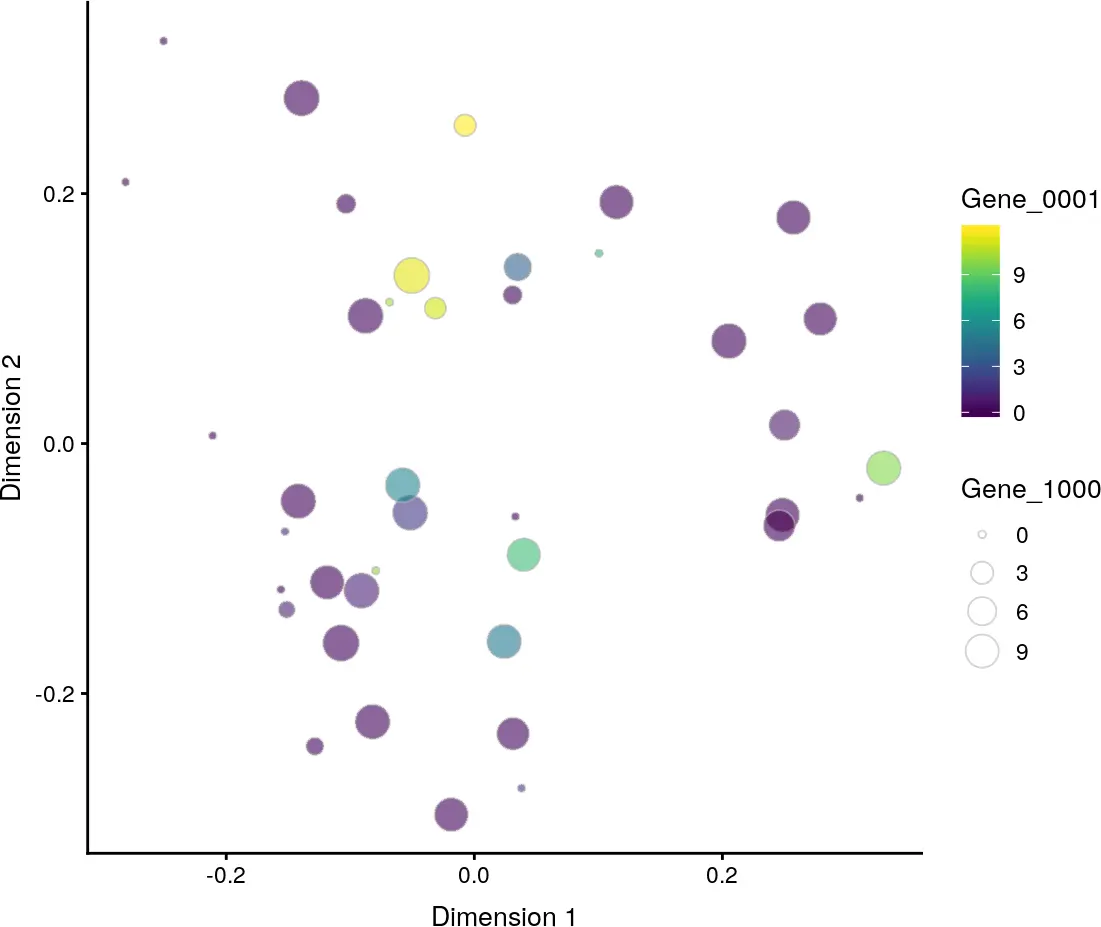
参考来源:http://www.bioconductor.org/packages/release/bioc/vignettes/scater/inst/doc/overview.html


若有收获,就点个赞吧
0 人点赞
