
PBMCs - 10X vs Microarray Reference
血液中的PBMCs是一种易于获取的异质细胞样品,其中包含了几种相似但截然不同的细胞类型。在10X Genomics官网中提供了一些数据集可供下载,如pbmc4k数据集,其中包含了来自健康个体的PBMC数据。此示例数据是用10X的cellRanger软件处理得到的,其中包括了几种不同的无监督聚类方法得到的细胞分群信息。
本示例将使用PBMCs细胞的参考数据来为这些聚类分群的细胞簇注释相应的生物细胞类型。Watkins等人(2009)已经发布了合适的PBMCs参考数据集(“HaemAtlas”)。他们分离纯化了PBMCs细胞类型的群体,并通过microarray测量了基因表达。此处使用的参考数据是从“haemosphere”网站下载的(Graaf等,2016)。
cell-ranger软件处理的数据产生了几种不同的细胞聚类分群结果,没有一个可能是完美的,但是将不同的聚类结果与类似的参考集进行比较可能有助于评估哪个聚类的结果最合适。此示例使用kmeans聚类的k=7的结果。
作为参考,以下是在cell-loupe browser中查看的t-SNE图分群的结果,不同细胞组标签后方括号中的数字是该组中的细胞数。
Prepare 10X query dataset
首先,将所需数据加载到SummarizedExperiment对象中,并过滤掉低表达基因或细胞成员过少的组。
# 加载celaref包library(celaref)datasets_dir <- "~/celaref_extra_vignette_data/datasets"# 使用load_dataset_10Xdata函数加载l0x cellRanger处理后的数据dataset_se.10X_pbmc4k_k7 <- load_dataset_10Xdata(dataset_path = file.path(datasets_dir,'10X_pbmc4k'),dataset_genome = "GRCh38",clustering_set = "kmeans_7_clusters",id_to_use = "GeneSymbol")# 使用trim_small_groups_and_low_expression_genes函数进行细胞和基因的过滤dataset_se.10X_pbmc4k_k7 <- trim_small_groups_and_low_expression_genes(dataset_se.10X_pbmc4k_k7)
然后,对过滤后的数据进行实验内不同组之间的差异比较分析。此处,将num-cores参数设置为7,以使每个组可以并行运行(指定较少以减少RAM使用)
# 使用contrast_each_group_to_the_rest函数进行差异分析
de_table.10X_pbmc4k_k7 <- contrast_each_group_to_the_rest(dataset_se.10X_pbmc4k_k7, dataset_name="10X_pbmc4k_k7", num_cores=7)
Prepare reference microarray dataset
接下来,我们对Watkins2009的参考数据集执行相同的操作。但是,由于这是microarray的数据,因此数据加载的过程有一些不同,并且实验内差异比较将使用contrast_each_group_to_the_rest_for_norm_ma_with_limma函数。该函数主要处理两件事:
- Logged, normalised expression values. Any low expression or poor quality measurements should have already been removed.
- Sample information.
this_dataset_dir <- file.path(datasets_dir, 'haemosphere_datasets','watkins')
# 表达矩阵路径
norm_expression_file <- file.path(this_dataset_dir, "watkins_expression.txt")
# 样本信息路径
samples_file <- file.path(this_dataset_dir, "watkins_samples.txt")
# 读取表达矩阵
norm_expression_table.full <- read.table(norm_expression_file, sep="\t", header=TRUE, quote="", comment.char="", row.names=1, check.names=FALSE)
# 读取样本信息
samples_table <- read_tsv(samples_file, col_types = cols())
samples_table$description <- make.names( samples_table$description)
# Avoid group or extra_factor names starting with numbers, for microarrays
从样本信息表中可以看到,该数据集中包含了其他组织,但是作为PBMCs的参考集,我们只希望考虑外周血的样本。因此,像其他数据加载功能一样,我们可以从中提取出所需的外周血样本的信息。
samples_table <- samples_table[samples_table$tissue == "Peripheral Blood",]
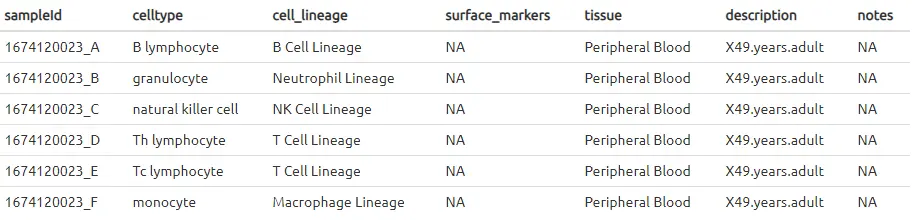
通常情况下,最困难的部分是格式化输入。参考集应使用与查询数据集中相同的基因ID,并将microarray的表达值进行log转换的归一化处理。
该数据来自Illumina HumanWG-6 v2 Expression BeadChips表达芯片,并在探针水平给出了基因的表达。这些探针需要转换为gene symbols以匹配PBMC数据。
注意:对于single cell datasets数据集,使用convert_se_gene_ids函数进行基因ID之间的转换更为容易。但是,该函数需要一个SummarizedExperiment对象,该对象不会用于microarray数据。因此,在此处,必须手动完成基因ID的匹配。
library("tidyverse")
library("illuminaHumanv2.db")
probes_with_gene_symbol_and_with_data <- intersect(keys(illuminaHumanv2SYMBOL),rownames(norm_expression_table.full))
# Get mappings - non NA
probe_to_symbol <- select(illuminaHumanv2.db, keys=rownames(norm_expression_table.full), columns=c("SYMBOL"), keytype="PROBEID")
probe_to_symbol <- unique(probe_to_symbol[! is.na(probe_to_symbol$SYMBOL),])
# no multimapping probes
genes_per_probe <- table(probe_to_symbol$PROBEID)
# How many genes a probe is annotated against?
multimap_probes <- names(genes_per_probe)[genes_per_probe > 1]
probe_to_symbol <- probe_to_symbol[!probe_to_symbol$PROBEID %in% multimap_probes, ]
convert_expression_table_ids <- function(expression_table, the_probes_table, old_id_name, new_id_name){
the_probes_table <- the_probes_table[,c(old_id_name, new_id_name)]
colnames(the_probes_table) <- c("old_id", "new_id")
# Before DE, just pick the top expresed probe to represent the gene
# Not perfect, but this is a ranking-based analysis.
# hybridisation issues aside, would expect higher epressed probes to be more relevant to Single cell data anyway.
probe_expression_levels <- rowSums(expression_table)
the_probes_table$avgexpr <- probe_expression_levels[as.character(the_probes_table$old_id)]
the_genes_table <- the_probes_table %>%
group_by(new_id) %>%
top_n(1, avgexpr)
expression_table <- expression_table[the_genes_table$old_id,]
rownames(expression_table) <- the_genes_table$new_id
return(expression_table)
}
# Just the most highly expressed probe foreach gene.
norm_expression_table.genes <- convert_expression_table_ids(
norm_expression_table.full,
probe_to_symbol,
old_id_name="PROBEID",
new_id_name="SYMBOL")
现在读取数据,并使用contrast_each_group_to_the_rest_for_norm_ma_with_limma函数进行实验内的差异比较分析。
由于在“description”字段中有关于每个样本来自哪个个体的信息,因此可以使用extra_factor_name参数进行指定,并将其作为线性模型中的一个因素包括在内。这是可选的,并且只能通过这种方式添加一个额外的因素。
# Go...
de_table.Watkins2009PBMCs <- contrast_each_group_to_the_rest_for_norm_ma_with_limma(
norm_expression_table = norm_expression_table.genes,
sample_sheet_table = samples_table,
dataset_name = "Watkins2009PBMCs",
extra_factor_name = 'description',
sample_name = "sampleId",
group_name = 'celltype')
Compare 10X query PBMCs to to reference
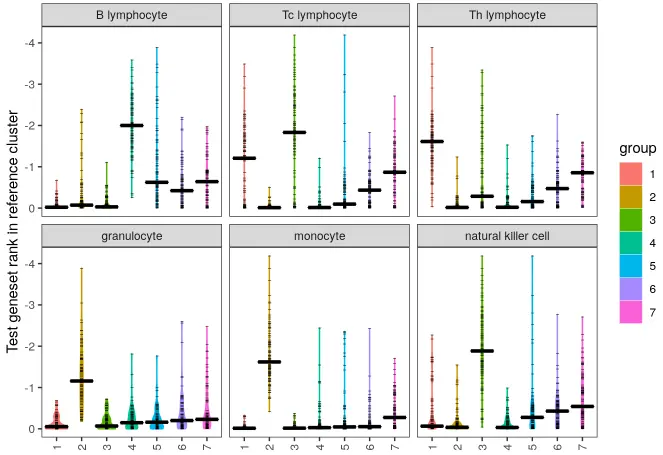
将查询数据集与参考集进行比较注释:
make_ranking_violin_plot(de_table.test = de_table.10X_pbmc4k_k7,
de_table.ref = de_table.Watkins2009PBMCs)
#> Warning: `fun.y` is deprecated. Use `fun` instead.
#> Warning: `fun.ymin` is deprecated. Use `fun.min` instead.
#> Warning: `fun.ymax` is deprecated. Use `fun.max` instead.
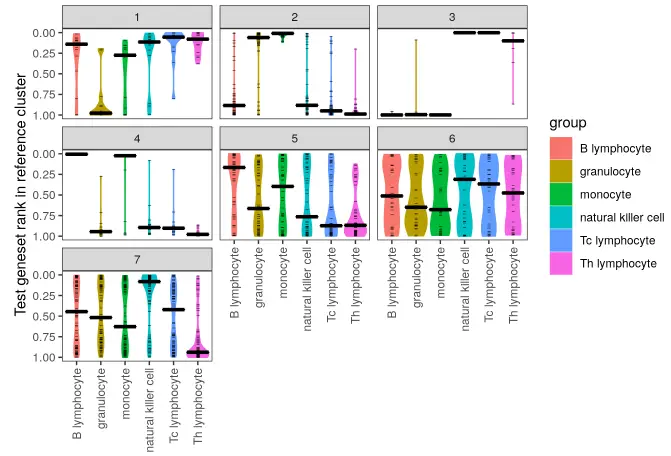
Hmm, there’s a few clusters where different the top genes are bunched near the top for a couple of different reference cell types.
Logging the plot will be more informative at the top end for this dataset.
make_ranking_violin_plot(de_table.test = de_table.10X_pbmc4k_k7,
de_table.ref = de_table.Watkins2009PBMCs, log10trans = TRUE)
#> Warning: `fun.y` is deprecated. Use `fun` instead.
#> Warning: `fun.ymin` is deprecated. Use `fun.min` instead.
#> Warning: `fun.ymax` is deprecated. Use `fun.max` instead.
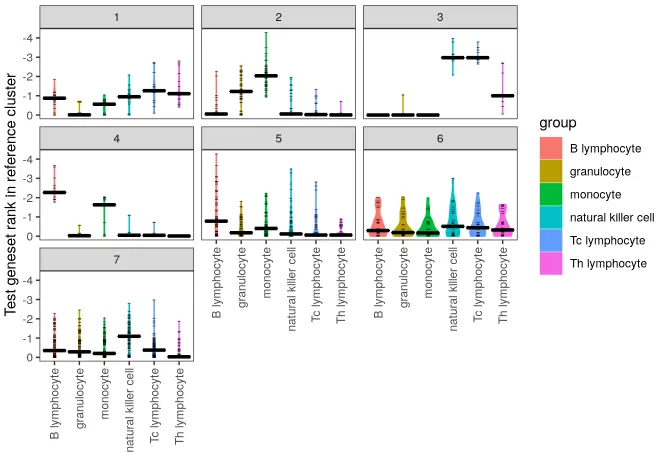
查看比较注释后的细胞分组标签:
label_table.pbmc4k_k7_vs_Watkins2009PBMCs <- make_ref_similarity_names(de_table.10X_pbmc4k_k7, de_table.Watkins2009PBMCs)
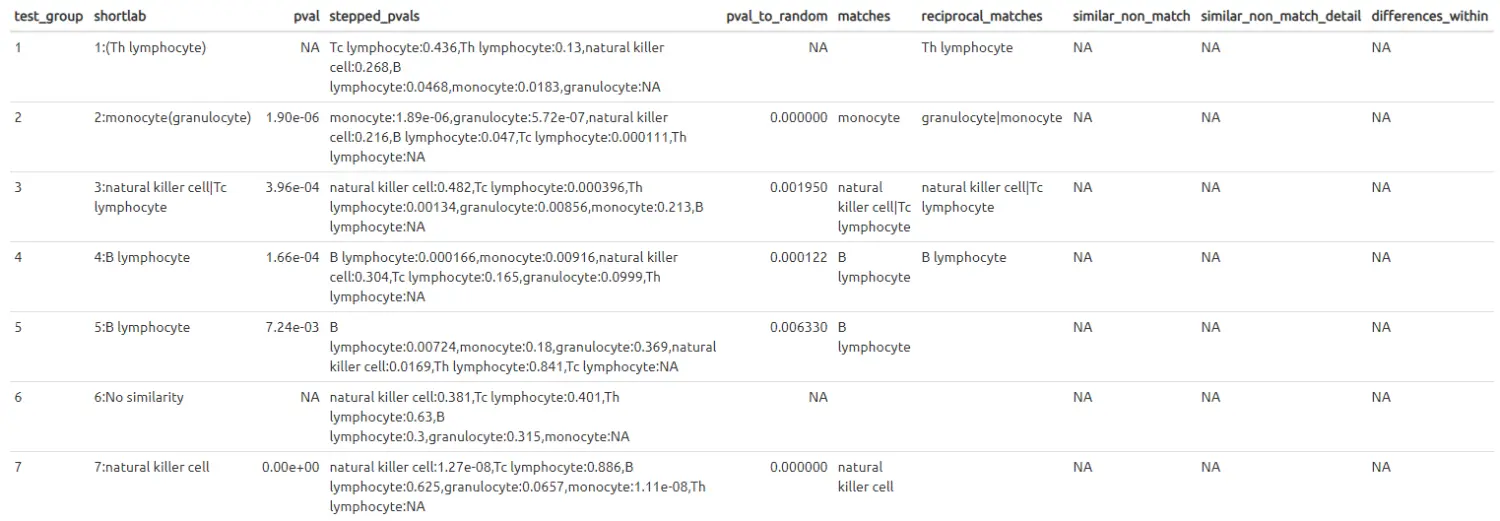
With a couple of (reciprocal-only matches) in the cluster names, it might be worth checking the reciprocal violin plots:
make_ranking_violin_plot(de_table.test = de_table.Watkins2009PBMCs,
de_table.ref = de_table.10X_pbmc4k_k7, log10trans = TRUE)
#> Warning: `fun.y` is deprecated. Use `fun` instead.
#> Warning: `fun.ymin` is deprecated. Use `fun.min` instead.
#> Warning: `fun.ymax` is deprecated. Use `fun.max` instead.
Mouse tissues - Similar and different
Zeisel等人(2015) 使用单细胞RNA-seq测序技术对小鼠的两个组织(sscortex和ca1hhicampus)进行了测序,揭示了小鼠皮质和海马体中的细胞类型。同样的,Farmer等人(2017)对小鼠泪腺两个不同发育阶段的细胞类型进行了调查,揭示了小鼠泪腺发育中上皮细胞的动态发育变化学和谱系关系。
这些细胞类型已经过了专业的描述,因此,我们可以将这些定义好的数据集作为参考数据集,与其他的单细胞数据集进行比较注释。
Load and compare mouse brain datasets
First, start by loading the brain cell data from (Zeisel et al. 2015):
datasets_dir <- "~/celaref_extra_vignette_data/datasets"
# 细胞样本信息
zeisel_cell_info_file <- file.path(datasets_dir, "zeisel2015", "zeisel2015_mouse_scs_detail.tab")
# 表达矩阵
zeisel_counts_file <- file.path(datasets_dir, "zeisel2015", "zeisel2015_mouse_scs_counts.tab")
请注意,zeisel2015_mouse_scs_detail.tab中的样本数据具有以下信息。它们在两个不同的水平上指定细胞类型组,对于本示例,我们仅使用level1class。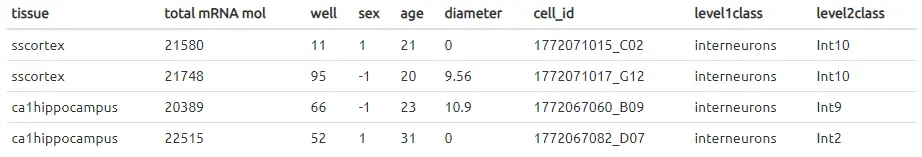
# 加载数据为SummarizedExperiment对象
dataset_se.zeisel <- load_se_from_files(zeisel_counts_file,
zeisel_cell_info_file,
group_col_name = "level1class",
cell_col_name = "cell_id" )
该dataset_se对象包含了所有数据,可以按组织将其分为两个对象(其为SummarizedExperiment对象)。然后分别过滤低表达基因和细胞数量少而无法分析的组。
# Subset the summarizedExperiment object into two tissue-specific objects
# 根据组织类型分成两个不同的对象
dataset_se.cortex <- dataset_se.zeisel[,dataset_se.zeisel$tissue == "sscortex"]
dataset_se.hippo <- dataset_se.zeisel[,dataset_se.zeisel$tissue == "ca1hippocampus"]
# And filter them
# 使用trim_small_groups_and_low_expression_genes函数对细胞和基因进行过滤
dataset_se.cortex <- trim_small_groups_and_low_expression_genes(dataset_se.cortex )
dataset_se.hippo <- trim_small_groups_and_low_expression_genes(dataset_se.hippo )
接下来,我们使用contrast_each_group_to_the_rest函数对数据集进行实验内的差异比较分析。每个样本中有6个细胞组,因此可以使用6个核心进行并行运算。这可能需要一些时间才能完成,因此请保存结果以供重新使用。
de_table.zeisel.cortex <- contrast_each_group_to_the_rest(dataset_se.cortex,
dataset_name="zeisel_sscortex",
num_cores=6)
de_table.zeisel.hippo <- contrast_each_group_to_the_rest(dataset_se.hippo,
dataset_name="zeisel_ca1hippocampus",
num_cores=6)
Now compare the two:
make_ranking_violin_plot(de_table.test = de_table.zeisel.cortex,
de_table.ref = de_table.zeisel.hippo)
#> Warning: `fun.y` is deprecated. Use `fun` instead.
#> Warning: `fun.ymin` is deprecated. Use `fun.min` instead.
#> Warning: `fun.ymax` is deprecated. Use `fun.max` instead.
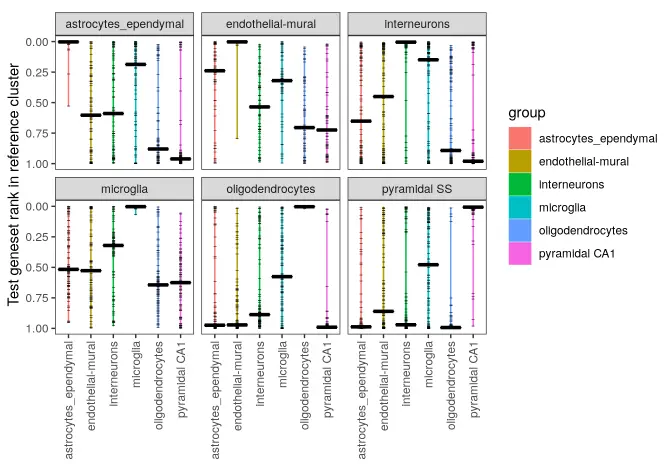
Perhaps unsurprisingly given they’re from the same experiment, the cell-type annotations do almost perfectly correlate one-to-one.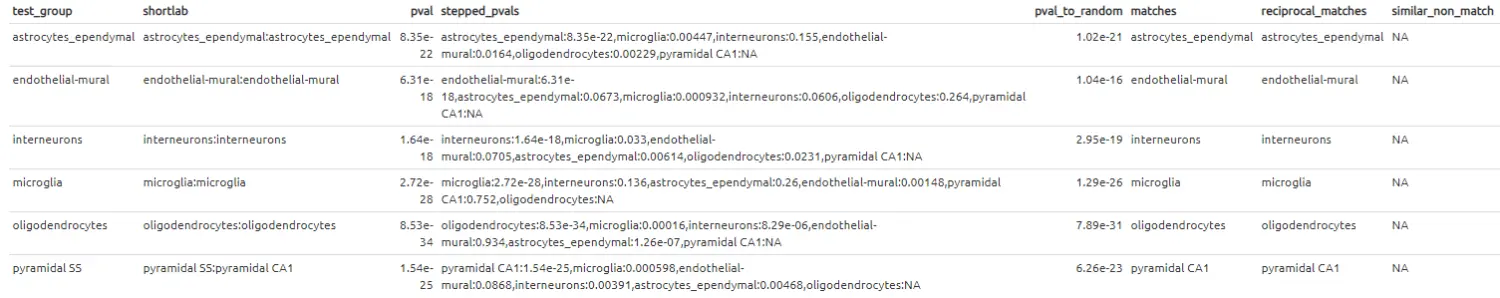
Load lacrimal gland dataset
接下来,我们将其与Farmer等人的泪腺(2017)数据集进行比较注释分析,这里仅使用更成熟的P4时间点的数据。该数据的格式稍微复杂一些,基因表达计数矩阵是一个MatrixMarket格式的文件,细胞分群信息位于单独的文件中。因此,我们需要将这些数据转换为load_se_from_tables函数期望的格式。
library(Matrix)
Farmer2017lacrimal_dir <- file.path(datasets_dir, "Farmer2017_lacrimal", "GSM2671416_P4")
# Counts matrix
Farmer2017lacrimal_matrix_file <- file.path(Farmer2017lacrimal_dir, "GSM2671416_P4_matrix.mtx")
Farmer2017lacrimal_barcodes_file <- file.path(Farmer2017lacrimal_dir, "GSM2671416_P4_barcodes.tsv")
Farmer2017lacrimal_genes_file <- file.path(Farmer2017lacrimal_dir, "GSM2671416_P4_genes.tsv")
counts_matrix <- readMM(Farmer2017lacrimal_matrix_file)
counts_matrix <- as.matrix(counts_matrix)
storage.mode(counts_matrix) <- "integer"
genes <- read.table(Farmer2017lacrimal_genes_file, sep="", stringsAsFactors = FALSE)[,1]
cells <- read.table(Farmer2017lacrimal_barcodes_file, sep="", stringsAsFactors = FALSE)[,1]
rownames(counts_matrix) <- genes
colnames(counts_matrix) <- cells
# Gene info table
gene_info_table.Farmer2017lacrimal <- as.data.frame(read.table(Farmer2017lacrimal_genes_file, sep="", stringsAsFactors = FALSE), stringsAsFactors = FALSE)
colnames(gene_info_table.Farmer2017lacrimal) <- c("ensemblID","GeneSymbol") # ensemblID is first, will become ID
## Cell/sample info
Farmer2017lacrimal_cells2groups_file <- file.path(datasets_dir, "Farmer2017_lacrimal", "Farmer2017_supps", paste0("P4_cellinfo.tab"))
Farmer2017lacrimal_clusterinfo_file <- file.path(datasets_dir, "Farmer2017_lacrimal", "Farmer2017_supps", paste0("Farmer2017_clusterinfo_P4.tab"))
# Cells to cluster number (just a number)
Farmer2017lacrimal_cells2groups_table <- read_tsv(Farmer2017lacrimal_cells2groups_file, col_types=cols())
# Cluster info - number to classification
Farmer2017lacrimal_clusterinfo_table <- read_tsv(Farmer2017lacrimal_clusterinfo_file, col_types=cols())
# Add in cluster info
Farmer2017lacrimal_cells2groups_table <- merge(x=Farmer2017lacrimal_cells2groups_table, y=Farmer2017lacrimal_clusterinfo_table, by.x="cluster", by.y="ClusterNum")
# Cell sample2group
cell_sample_2_group.Farmer2017lacrimal <- Farmer2017lacrimal_cells2groups_table[,c("Cell identity","ClusterID", "nGene", "nUMI")]
colnames(cell_sample_2_group.Farmer2017lacrimal) <- c("cell_sample", "group", "nGene", "nUMI")
# Add -1 onto each of the names, that seems to be in the counts
cell_sample_2_group.Farmer2017lacrimal$cell_sample <- paste0(cell_sample_2_group.Farmer2017lacrimal$cell_sample, "-1")
# Create a summarised experiment object.
dataset_se.P4 <- load_se_from_tables(counts_matrix,
cell_info_table = cell_sample_2_group.Farmer2017lacrimal,
gene_info_table = gene_info_table.Farmer2017lacrimal )
After all that, the dataset has the cell information (colData):
… and the gene information (rowData):
请注意,“ID”列是ensembl的基因ID,我们需要将其转换为gene symbol,以匹配Zeisel的数据。Gene symbol和ID之间的转换几乎是一对一的映射关系,因此,在此步骤中可能会丢失了一些对应不上的基因。
rowData(dataset_se.P4)$total_count <- rowSums(assay(dataset_se.P4))
# 使用convert_se_gene_ids函数进行基因ID转换
dataset_se.P4 <- convert_se_gene_ids( dataset_se.P4, new_id='GeneSymbol', eval_col='total_count')
转换好基因ID后,我们将过滤掉低表达的基因和较小的细胞组,并进行实验内的差异比较分析(within-experiment comparisons):
# 使用trim_small_groups_and_low_expression_genes函数进行细胞和基因的过滤
dataset_se.P4 <- trim_small_groups_and_low_expression_genes(dataset_se.P4)
# 使用contrast_each_group_to_the_rest函数进行差异比较分析
de_table.Farmer2017lacrimalP4 <- contrast_each_group_to_the_rest(
dataset_se.P4,
dataset_name="Farmer2017lacrimalP4",
num_cores = 4)
Cross-tissue comparision
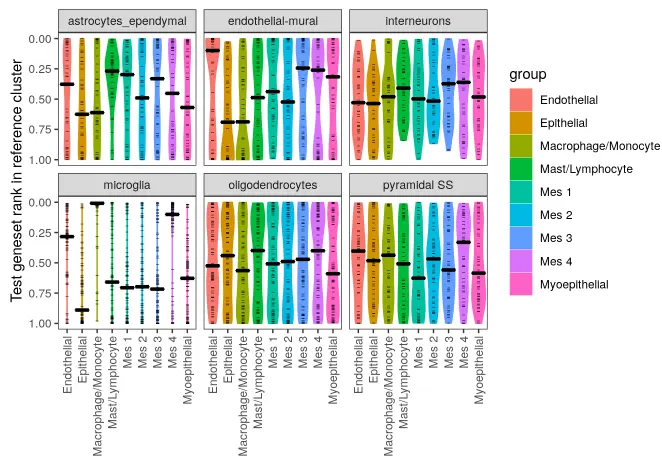
现在,我们将小鼠cortex样本与泪腺(lacrimal gland)数据进行比较注释。作为完全不同的组织,应该没有太多共同的细胞类型。
make_ranking_violin_plot(de_table.test = de_table.zeisel.cortex,
de_table.ref = de_table.Farmer2017lacrimalP4)
#> Warning: `fun.y` is deprecated. Use `fun` instead.
#> Warning: `fun.ymin` is deprecated. Use `fun.min` instead.
#> Warning: `fun.ymax` is deprecated. Use `fun.max` instead.
# 查看比较注释的结果
label_table.cortex_vs_lacrimal <- make_ref_similarity_names(
de_table.zeisel.cortex,
de_table.Farmer2017lacrimalP4)
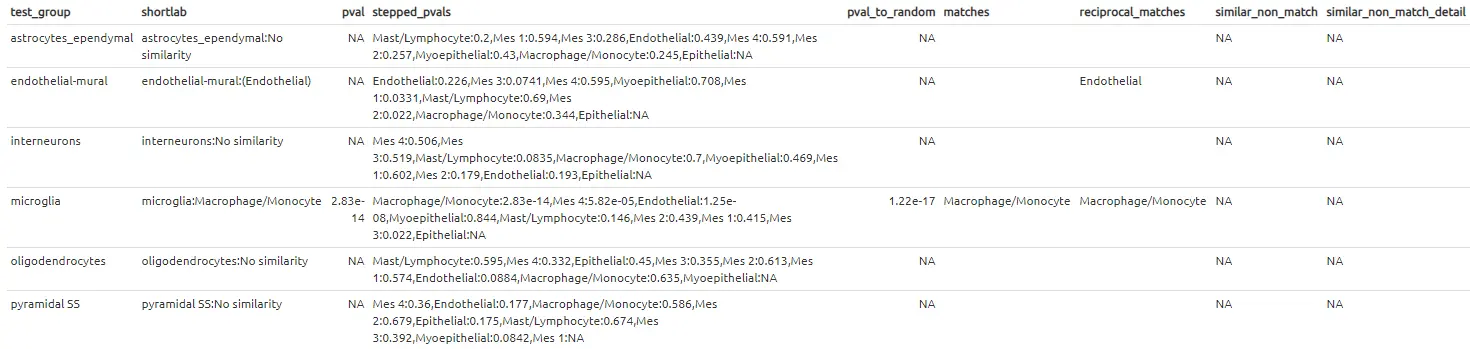
这种跨组织的比较结果看起来与上面大脑和大脑之间的对比结果非常的不同-可以预期,大多数的细胞簇没有“相似性”。虽然不是全部-大脑皮层中的“microglia(小胶质细胞)”组与泪腺数据集中的“Macrophage/Monocyte(巨噬/单核细胞)”组有相似之处。这是有道理的,因为它们是生物学上相似的细胞类型。有趣的是,大脑样本中的内皮壁细胞(endothelial-mural cell)与泪腺中的内皮细胞(endothelial cells)之间也存在着相互的匹配。
参考来源:http://www.bioconductor.org/packages/release/bioc/vignettes/celaref/inst/doc/celaref_doco.html

若有收获,就点个赞吧
0 人点赞
