Signac是Seurat的一个扩展功能R包,可以用来分析、解释和探索
单细胞染色质数据集。目前,Signac包主要专注于分析单细胞ATAC-seq数据,之后还会添加其他基于染色质调控的单细胞测序数据分析的新功能。Signac包主要支持以下功能:
- 单细胞数据质控指标的计算
- 数据降维、可视化和细胞聚类
- 细胞类型特异性peak的鉴定
- “pseudo-bulk” coverage tracks的可视化
- 多样本scATAC-seq数据集的整合
- 与scRNA-seq数据集的整合
- Motif富集分析
本教程中,我们将学习处理10x Genomics平台测序产生的人外周血单核细胞(PBMC)的scATAC-seq数据,原始的示例数据可以从10x Genomics官网进行下载:
- The Raw data
- The Metadata
- The fragments file
- The fragments file index
安装Signac及其依赖包
# 安装bioconductor包# Install bioconductorif (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install()# 安装Signac包# Current releaseinstall.packages("Signac")# Development versioninstall.packages("devtools")devtools::install_github("timoast/signac", ref = "develop")#安装参考基因组的注释信息# HumanBiocManager::install(c('BSgenome.Hsapiens.UCSC.hg19', 'EnsDb.Hsapiens.v75'))# MouseBiocManager::install(c('BSgenome.Mmusculus.UCSC.mm10', 'EnsDb.Mmusculus.v79'))
加载所需的R包
library(Signac)library(Seurat)library(GenomeInfoDb)library(EnsDb.Hsapiens.v75)library(ggplot2)library(patchwork)set.seed(1234)
数据加载与预处理
Signac主要读取两个输入文件,它们可以用CellRanger处理产生:
- Peak/Cell matrix.
它类似于用于分析单细胞RNA-seq的基因表达计数矩阵。但是,矩阵中的每一行代表一个基因组区域(“peak region”)而不是基因,表示开放的染色质区域。矩阵中的每个值代表在每个峰内映射的每个条形码(即细胞)的Tn5切割位点的数量,我们可以在10X Genomics官网上找到更多详细的信息。 - Fragment file.
这个文件存储了所有单细胞中唯一片段(unique fragments)的完整列表。该文件较大,使用起来也较慢,并且存储在磁盘上(而不是内存中)。但是,保留此文件的好处是它包含了与每个细胞关联的所有片段,与仅映射到peaks的reads数相反。
对于大多数分析,我们仅使用peak/cell matrix矩阵即可,但对于某些分析(如,创建“Gene Activity Matrix”),我们发现仅使用peaks映射的reads数可能会对结果产生不利的影响。因此,在本教程中,我们同时使用了这两个文件。首先,我们使用peak/cell matrix矩阵创建Seurat对象,然后将片段文件(fragment file)的路径存储在磁盘上的Seurat对象中:
# 读取peak/cell matrix矩阵
counts <- Read10X_h5(filename = "/home/dongwei/scATAC-seq/PBMC/atac_v1_pbmc_10k_filtered_peak_bc_matrix.h5")
# 查看counts信息
head(counts)
6 x 9277 sparse Matrix of class "dgCMatrix"
[[ suppressing 25 column names 'AAACGAAAGAGCGAAA-1', 'AAACGAAAGAGTTTGA-1', 'AAACGAAAGCGAGCTA-1' ... ]]
chr1:565113-565543 . . . . . . . . . . . . . . . . . . . . . .
chr1:569179-569635 . . . . . . . . . . . . . . . . . . . . . .
chr1:713534-714806 . . 2 8 . 2 2 . . 2 . . . . . 4 . . . . . .
chr1:752436-753020 . . . . . . . . . . . . . . . . . . 1 . . .
chr1:762144-763353 . . 4 2 . . . 2 . . . . . . 2 . 2 . 4 . . .
chr1:779610-780252 . . . . . . . . . . . . . . . . . . . . . .
chr1:565113-565543 . . . ......
chr1:569179-569635 . . . ......
chr1:713534-714806 2 . . ......
chr1:752436-753020 . 2 . ......
chr1:762144-763353 . 2 2 ......
chr1:779610-780252 . . 1 ......
.....suppressing 9252 columns in show(); maybe adjust 'options(max.print= *, width = *)'
..............................
# 查看peaks和细胞的数目
dim(counts)
[1] 80234 9277
# 读取细胞注释信息
metadata <- read.csv(file = "/home/dongwei/scATAC-seq/PBMC/atac_v1_pbmc_10k_singlecell.csv",
header = TRUE,
row.names = 1
)
# 查看metadata信息
names(metadata)
[1] "total"
[2] "duplicate"
[3] "chimeric"
[4] "unmapped"
[5] "lowmapq"
[6] "mitochondrial"
[7] "passed_filters"
[8] "cell_id"
[9] "is__cell_barcode"
[10] "TSS_fragments"
[11] "DNase_sensitive_region_fragments"
[12] "enhancer_region_fragments"
[13] "promoter_region_fragments"
[14] "on_target_fragments"
[15] "blacklist_region_fragments"
[16] "peak_region_fragments"
[17] "peak_region_cutsites"
head(metadata)[,1:5]
total duplicate chimeric unmapped lowmapq
NO_BARCODE 9034214 3902029 106211 1424804 648333
AAACGAAAGAAACGCC-1 3 1 0 1 0
AAACGAAAGAAAGCAG-1 15 0 0 5 3
AAACGAAAGAAAGGGT-1 8 1 0 2 0
AAACGAAAGAAATACC-1 9908 5343 120 121 1146
AAACGAAAGAAATCTG-1 2 0 0 0 0
# 构建Seurat对象
pbmc <- CreateSeuratObject(
counts = counts,
assay = 'peaks',
project = 'ATAC',
min.cells = 1,
meta.data = metadata
)
pbmc
An object of class Seurat
79921 features across 9277 samples within 1 assay
Active assay: peaks (79921 features, 0 variable features)
# 指定fragment file路径
fragment.path <- '/home/dongwei/scATAC-seq/PBMC/atac_v1_pbmc_10k_fragments.tsv.gz'
# 使用SetFragments函数设置fragment file路径
pbmc <- SetFragments(
object = pbmc,
file = fragment.path
)
# 查看Seurat对象
pbmc
An object of class Seurat
79921 features across 9277 samples within 1 assay
Active assay: peaks (79921 features, 0 variable features)
Optional step: Filtering the fragment file
To make downstream steps that use this file faster, we can filter the fragments file to contain only reads from cells that we retain in the analysis. This is optional, but slow, and only needs to be performed once. Running this command writes a new file to disk and indexes the file so it is ready to be used by Signac.
# 指定过滤后的fragment file
fragment_file_filtered <- "/home/dongwei/scATAC-seq/PBMC/atac_v1_pbmc_10k_filtered_fragments.tsv"
# 使用FilterFragments函数对fragment file进行过滤
FilterFragments(
fragment.path = fragment.path,
cells = colnames(pbmc),
output.path = fragment_file_filtered
)
Retaining 9277 cells
Reading fragments
|--------------------------------------------------|
|==================================================|
Sorting fragments
Writing output
Compressing output
Building index
# 使用过滤后的fragment file重新设置路径
pbmc <- SetFragments(object = pbmc, file = paste0(fragment_file_filtered, '.bgz'))
计算QC指标进行数据质控
目前,对于scATAC-seq测序数据,我们建议使用以下QC指标来评估数据质量。与scRNA-seq数据一样,这些参数的预期值范围将取决于我们的生物系统,细胞生存力等。
- Nucleosome banding pattern(核小体条带模式):绘制片段大小(fragment sizes)的直方图(由配对末端测序读数确定)来展示核小体的条带模式。我们计算每个单细胞,并量化单核小体与无核小体片段(存储为nucleosome_signal)的近似比率。
- Transcriptional start site (TSS) enrichment score(转录起始位点富集得分):
ENCODE项目已根据以TSS为中心的片段与TSS侧翼区域中的片段的比率定义了ATAC-seq定位得分(请参阅https://www.encodeproject.org/data-standards/terms/)。较差的ATAC-seq实验通常将具有较低的TSS富集得分。我们可以使用TSSEnrichment函数为每个细胞计算该指标,并将结果存储在元数据中,列名称为TSS.enrichment。 - Total number of fragments in peaks(peaks中片段的总数):细胞测序深度/复杂性的量度。由于测序深度低,可能需要排除reads数很少的细胞,reads数极高的细胞可能代表了doublets或核团块。
- Fraction of fragments in peaks(peaks中片段的比例):表示落在ATAC-seq peaks内的总片段比例。比例较低的细胞(即<15-20%)通常代表应删除的低质量细胞或技术误差。
- Ratio reads in ‘blacklist’ sites(比对到“blacklist”区域的reads比率):
ENCODE项目提供了一个“blacklist“”区域列表,这些区域表示通常与人为信号关联的读取。reads比对到这些区域的比例较高的细胞(与reads比对到peaks区域比例相比)通常代表了技术误差,应将其删除。Signac包中包含了针对人类(hg19和GRCh38),小鼠(mm10),果蝇(dm3)和秀丽隐杆线虫(ce10)的ENCODE“blacklist”区域。
# 使用NucleosomeSignal函数计算Nucleosome banding pattern
pbmc <- NucleosomeSignal(object = pbmc)
# 计算peaks中reads的比例
pbmc$pct_reads_in_peaks <- pbmc$peak_region_fragments / pbmc$passed_filters * 100
# 计算比对到“blacklist”区域的reads比率
pbmc$blacklist_ratio <- pbmc$blacklist_region_fragments / pbmc$peak_region_fragments
# 质控指标的可视化
p1 <- VlnPlot(pbmc, c('pct_reads_in_peaks', 'peak_region_fragments'), pt.size = 0.1)
p2 <- VlnPlot(pbmc, c('blacklist_ratio', 'nucleosome_signal'), pt.size = 0.1) & scale_y_log10()
p1 | p2
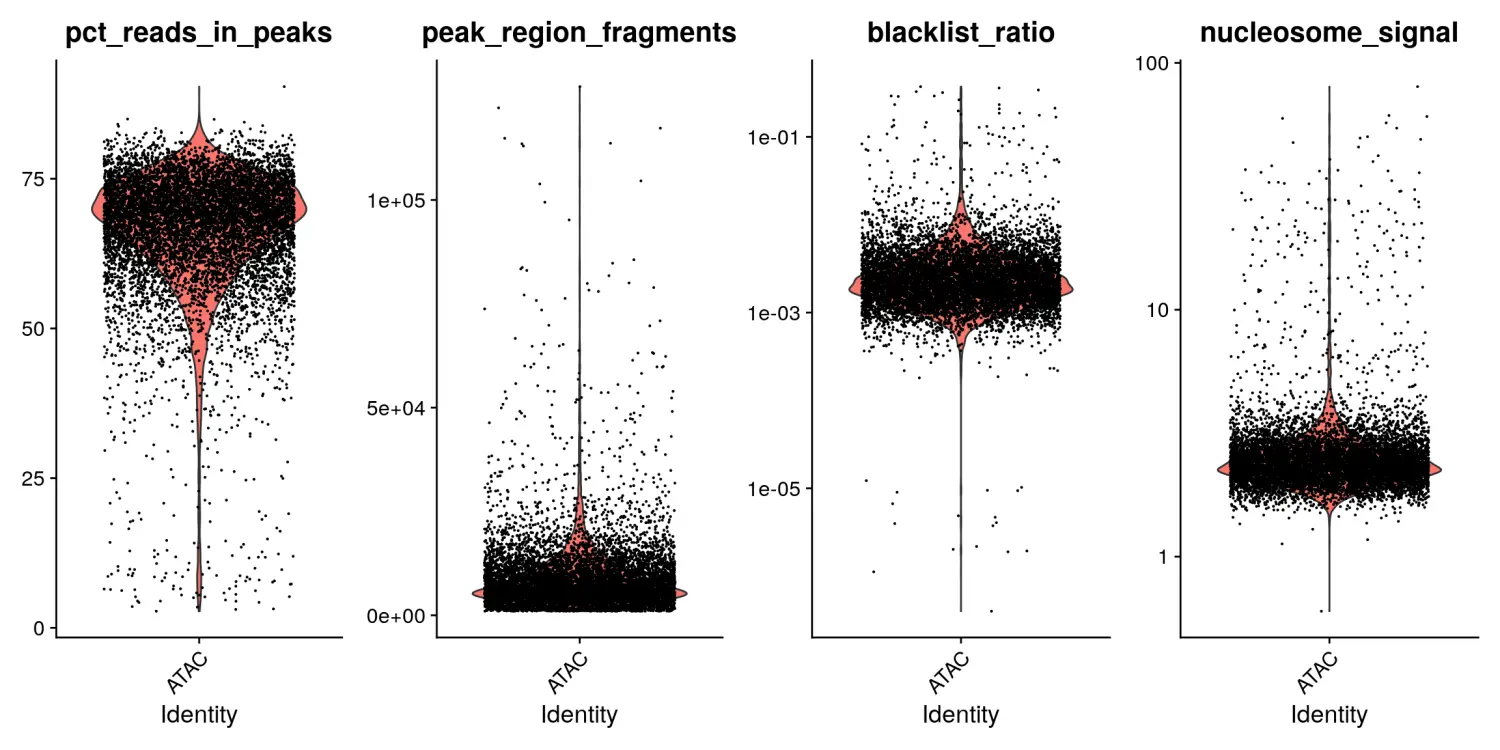
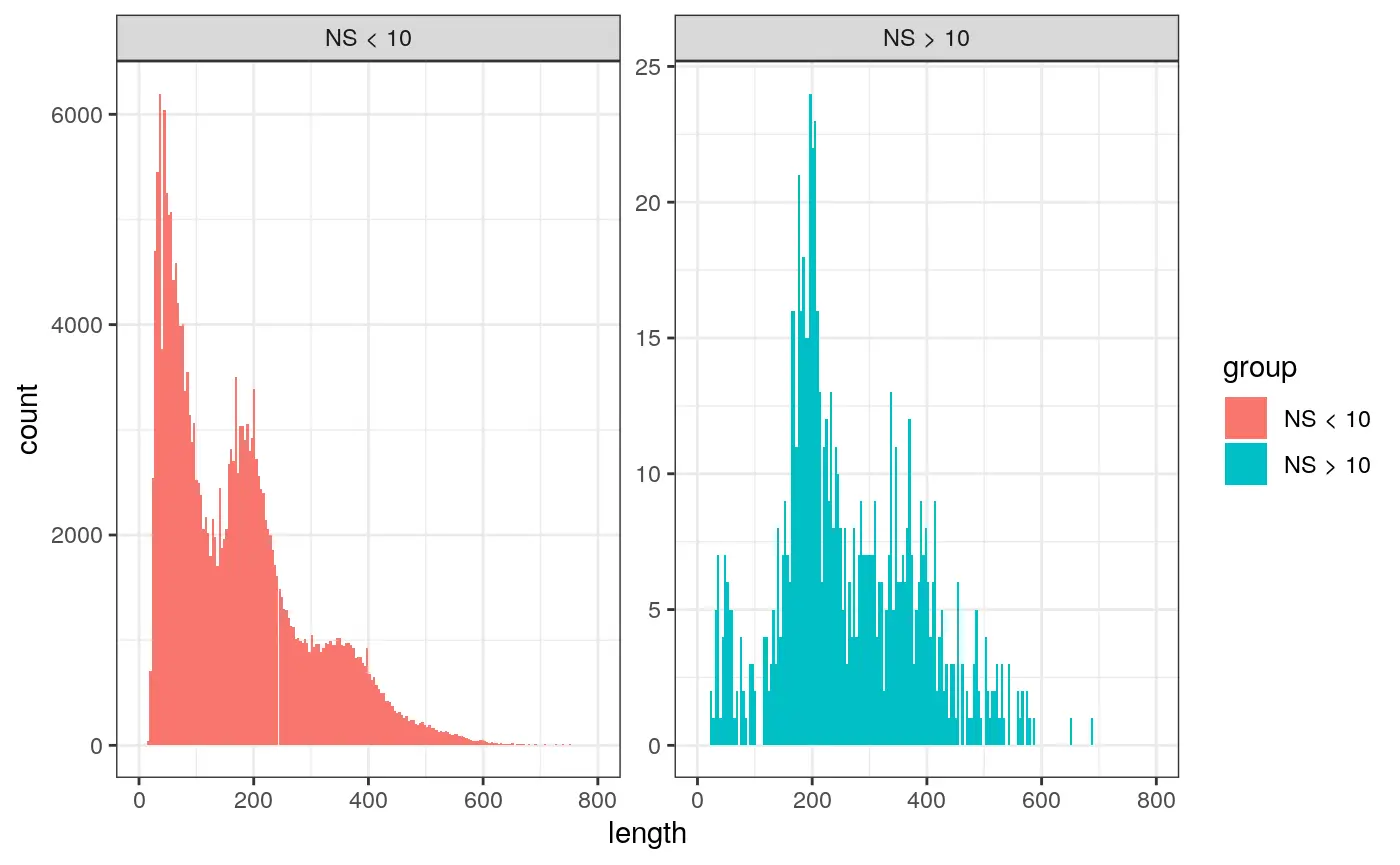
我们还可以查看所有细胞的片段长度的周期性,并按具有高或低的核小体信号强度对细胞进行分组。可以发现,与单核小体/无核小体比率不同的细胞具有不同的核小体条带模式。
# 根据核小体条带信号强度进行分组
pbmc$nucleosome_group <- ifelse(pbmc$nucleosome_signal > 10, 'NS > 10', 'NS < 10')
# 使用FragmentHistogram函数可视化核小体条带分布模式
FragmentHistogram(object = pbmc, group.by = 'nucleosome_group')
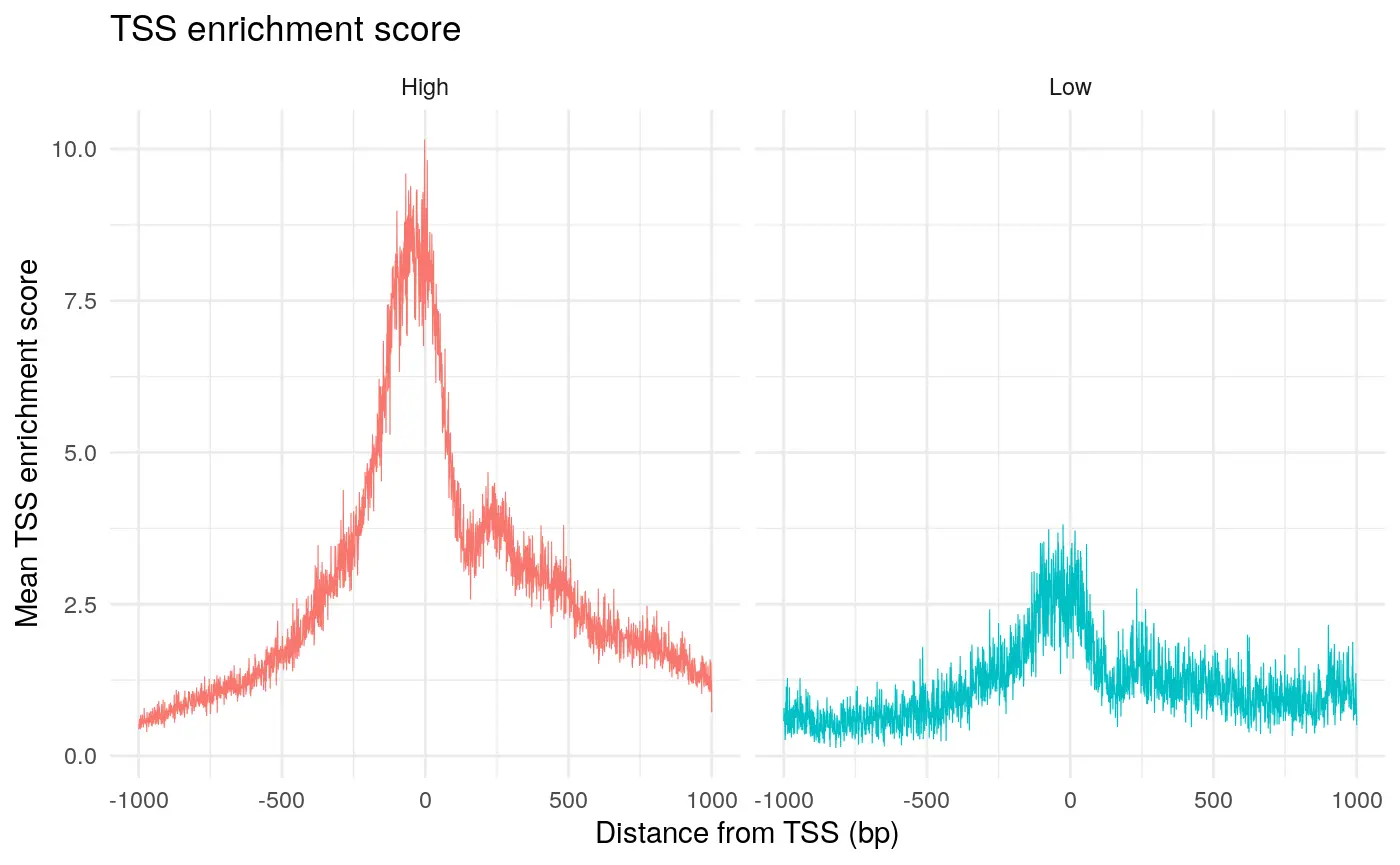
Tn5整合事件在转录起始位点(TSS)的富集也是评估ATAC-seq实验中Tn5靶向的重要质量控制指标。ENCODE联盟将TSS富集分数定义为将TSS周围的Tn5整合位点数量标准化为侧翼区域中Tn5整合位点的数量。有关TSS富集得分的更多信息,请参阅ENCODE文档(https://www.encodeproject.org/data-standards/terms/)。我们可以使用Signac包中的`TSSEnrichment`函数来计算每个细胞的TSS富集得分。
# create granges object with TSS positions
gene.ranges <- genes(EnsDb.Hsapiens.v75)
seqlevelsStyle(gene.ranges) <- 'UCSC'
gene.ranges <- gene.ranges[gene.ranges$gene_biotype == 'protein_coding', ]
gene.ranges <- keepStandardChromosomes(gene.ranges, pruning.mode = 'coarse')
tss.ranges <- GRanges(
seqnames = seqnames(gene.ranges),
ranges = IRanges(start = start(gene.ranges), width = 2),
strand = strand(gene.ranges)
)
seqlevelsStyle(tss.ranges) <- 'UCSC'
tss.ranges <- keepStandardChromosomes(tss.ranges, pruning.mode = 'coarse')
# to save time use the first 2000 TSSs
# 使用TSSEnrichment函数计算TSS富集得分
pbmc <- TSSEnrichment(object = pbmc, tss.positions = tss.ranges[1:2000])
pbmc$high.tss <- ifelse(pbmc$TSS.enrichment > 2, 'High', 'Low')
# 使用TSSPlot函数对TSS富集得分进行可视化
TSSPlot(pbmc, group.by = 'high.tss') + ggtitle("TSS enrichment score") + NoLegend()
# Finally we remove cells that are outliers for these QC metrics.
# 根据不同QC指标进行数据过滤
pbmc <- subset(
x = pbmc,
subset = peak_region_fragments > 3000 &
peak_region_fragments < 20000 &
pct_reads_in_peaks > 15 &
blacklist_ratio < 0.05 &
nucleosome_signal < 10 &
TSS.enrichment > 2
)
pbmc
## An object of class Seurat
## 89307 features across 6978 samples within 1 assay
## Active assay: peaks (89307 features, 0 variable features)
数据归一化与线性降维
- Normalization(数据归一化): Signac包使用
frequency-inverse document frequency (TF-IDF)方法进行数据归一化处理。它将进行两步归一化的过程,既可以跨细胞进行归一化以校正细胞测序深度的差异,也可以跨峰(peaks)进行归一化为更罕见的峰提供更高的值。 - Feature selection(特征选择): 正如我们对scRNA-seq数据所做的那样,scATAC-seq数据的大部分二进制性质(binary nature)使其难以执行“可变”特征选择。取而代之的是,我们可以选择仅使用前n%个特征(峰)进行数据降维,或者使用
FindTopFeatures函数删除少于在n个细胞中出现的峰。这里,我们将使用所有的peaks,尽管我们注意到,仅使用部分features(尝试将min.cutoff设置为“q75”以使用前25%的峰)会看到非常相似的结果,并且运行速度更快。通过此功能,用于数据降维的特征将自动被存储为Seurat对象中的VariableFeatures。 - Dimensional reduction(数据降维): 接下来,我们使用上面选择的特征(峰)在TD-IDF归一化矩阵上运行奇异值分解(SVD),最后返回对象的低维数据表示结果(对于熟悉scRNA-seq数据的用户,您可以认为这类似于PCA的输出)。
# 使用RunTFIDF函数进行数据归一化
pbmc <- RunTFIDF(pbmc)
# 使用FindTopFeatures函数选择可变的features
pbmc <- FindTopFeatures(pbmc, min.cutoff = 'q0')
# 使用RunSVD函数进行线性降维
pbmc <- RunSVD(
object = pbmc,
assay = 'peaks',
reduction.key = 'LSI_',
reduction.name = 'lsi'
)
线性降维后,第一个LSI成分通常捕获测序深度(技术差异),而不是生物学差异。在这种情况下,应从下游分析中删除该成分。我们可以使用DepthCor函数评估每个LSI成分与测序深度之间的相关性:
DepthCor(pbmc)
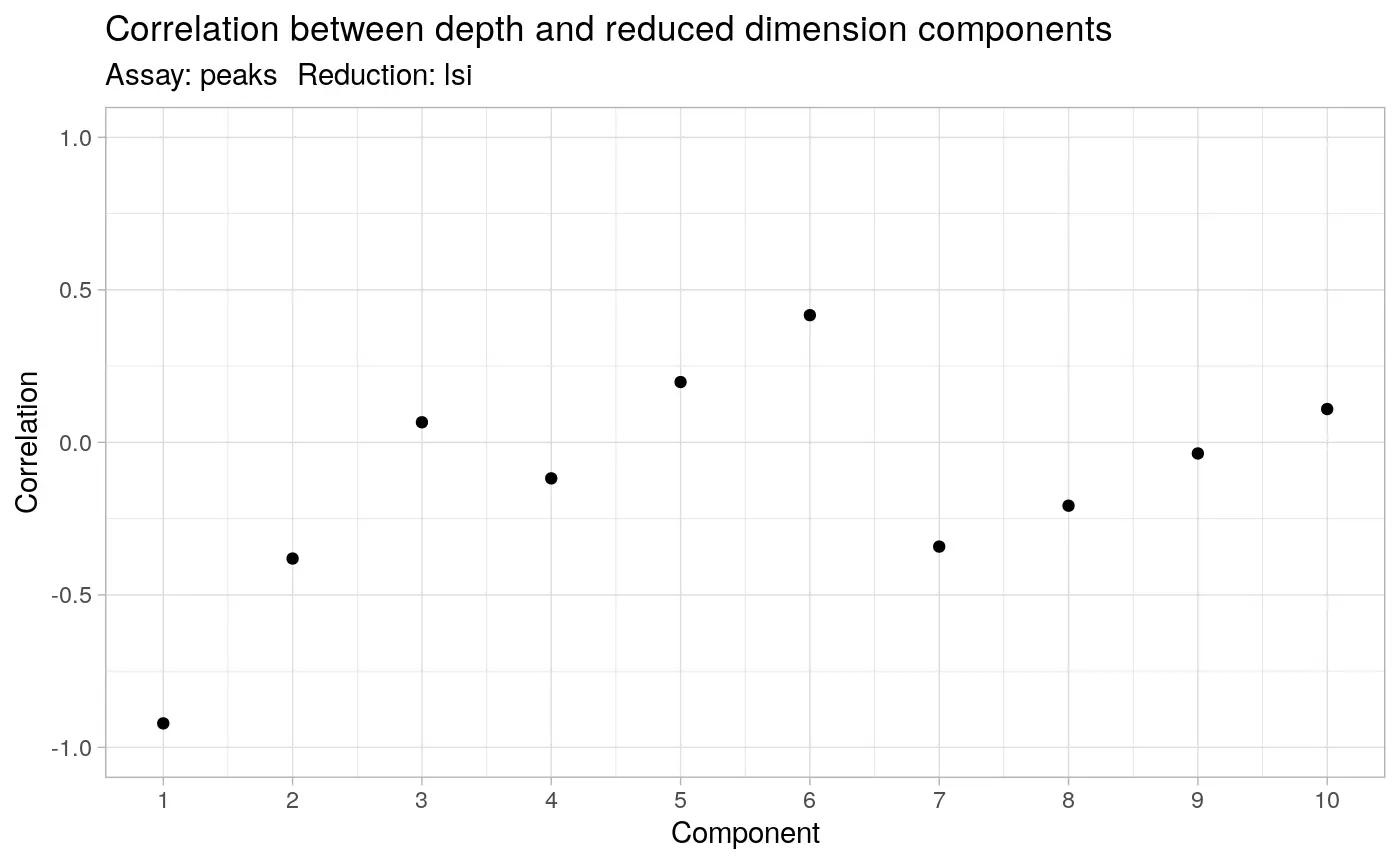
从上图中,我们可以看到第一个LSI成分与细胞中counts的总数之间有非常强的相关性,因此我们将在后续的分析中删除此成分。
非线性降维与细胞聚类
数据线性降维后,我们可以使用通常用于分析scRNA-seq数据的方法来对细胞进行基于图的聚类,并进行非线性降维(如UMAP等)可视化。其中,函数RunUMAP,FindNeighbors和FindClusters都来自Seurat包。
# 使用RunUMAP函数进行UMAP非线性降维
pbmc <- RunUMAP(object = pbmc, reduction = 'lsi', dims = 2:30)
# 对细胞执行基于图的聚类
pbmc <- FindNeighbors(object = pbmc, reduction = 'lsi', dims = 2:30)
pbmc <- FindClusters(object = pbmc, verbose = FALSE, algorithm = 3)
# 使用DimPlot函数进行数据可视化
DimPlot(object = pbmc, label = TRUE) + NoLegend()
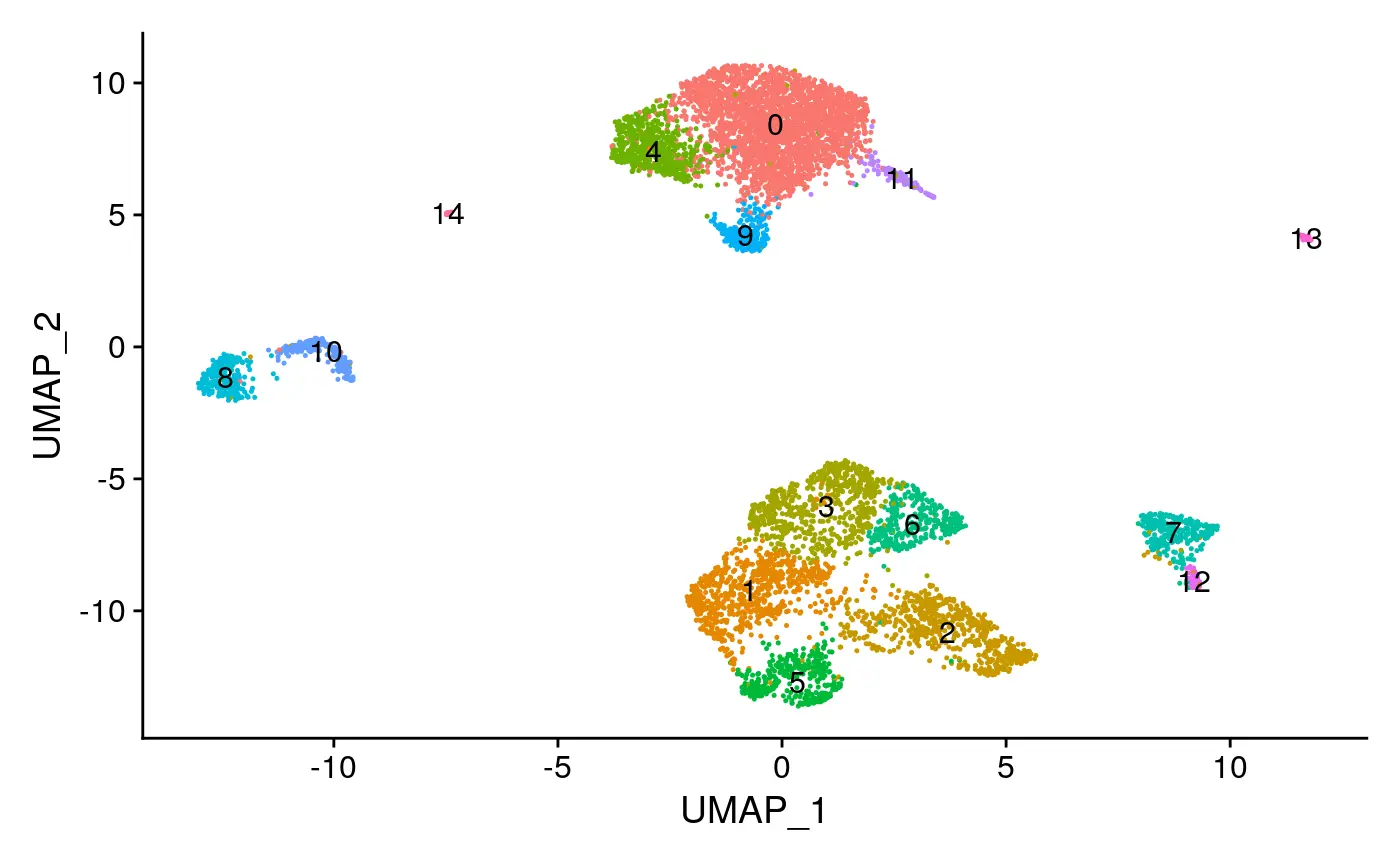
创建基因表达活性矩阵(gene activity matrix)
从上图UMAP降维可视化的结果中可以看出,在人的外周血中存在多个细胞类群。如果我们还对PBMC的scRNA-seq分析结果熟悉的话,甚至还可以在scATAC-seq数据中发现某些髓样和淋巴样细胞群体的存在。但是,在scATAC-seq数据中注释和解释不同的细胞簇更具有挑战性,因为我们对非编码基因组区域功能的了解远少于对蛋白质编码区域(基因)的了解。
但是,我们可以尝试通过评估与每个基因相关的染色质可及性来量化基因组中每个基因的表达活性,并创建一个基于scATAC-seq数据的新基因活性测定方法。在这里,我们将使用一种简单的方法来对与基因体和启动子区域相交的reads数进行求和(我们也建议您探索Cicero工具,它可以实现类似的目标)。
为了创建基因表达活性矩阵(gene activity matrix),我们从EnsembleDB中提取了人类基因组的基因坐标,并将其扩展到TSS上游2kb区域(因为启动子区域的可及性通常与基因表达相关)。然后,我们使用FeatureMatrix函数计算映射到这些区域的每个细胞的片段数。这将获取任何一组基因组坐标,计算与每个细胞中的这些坐标相交的reads次数,并返回一个稀疏矩阵。
# Extend coordinates upstream to include the promoter
genebodyandpromoter.coords <- Extend(x = gene.ranges, upstream = 2000, downstream = 0)
# create a gene by cell matrix
gene.activities <- FeatureMatrix(
fragments = fragment.path,
features = genebodyandpromoter.coords,
cells = colnames(pbmc),
chunk = 20
)
# convert rownames from chromsomal coordinates into gene names
gene.key <- genebodyandpromoter.coords$gene_name
names(gene.key) <- GRangesToString(grange = genebodyandpromoter.coords)
rownames(gene.activities) <- gene.key[rownames(gene.activities)]
# add the gene activity matrix to the Seurat object as a new assay, and normalize it
pbmc[['RNA']] <- CreateAssayObject(counts = gene.activities)
pbmc <- NormalizeData(
object = pbmc,
assay = 'RNA',
normalization.method = 'LogNormalize',
scale.factor = median(pbmc$nCount_RNA)
)
现在,我们可以对一些典型的marker基因的“活性”进行可视化展示,以帮助我们解释scATAC-seq数据聚类分出的不同细胞簇。请注意,该基因“活性”会比scRNA-seq数据测量值的噪音大得多。这是因为它们代表了来自稀疏染色质数据的预测值,而且还因为它们假定了基因体/启动子可及性与基因表达之间的一般对应关系,而实际情况并非总是如此。尽管如此,我们仍可以基于此数据识别出单核细胞,B细胞,T细胞和NK细胞的不同类群。
DefaultAssay(pbmc) <- 'RNA'
FeaturePlot(
object = pbmc,
features = c('MS4A1', 'CD3D', 'LEF1', 'NKG7', 'TREM1', 'LYZ'),
pt.size = 0.1,
max.cutoff = 'q95',
ncol = 3
)
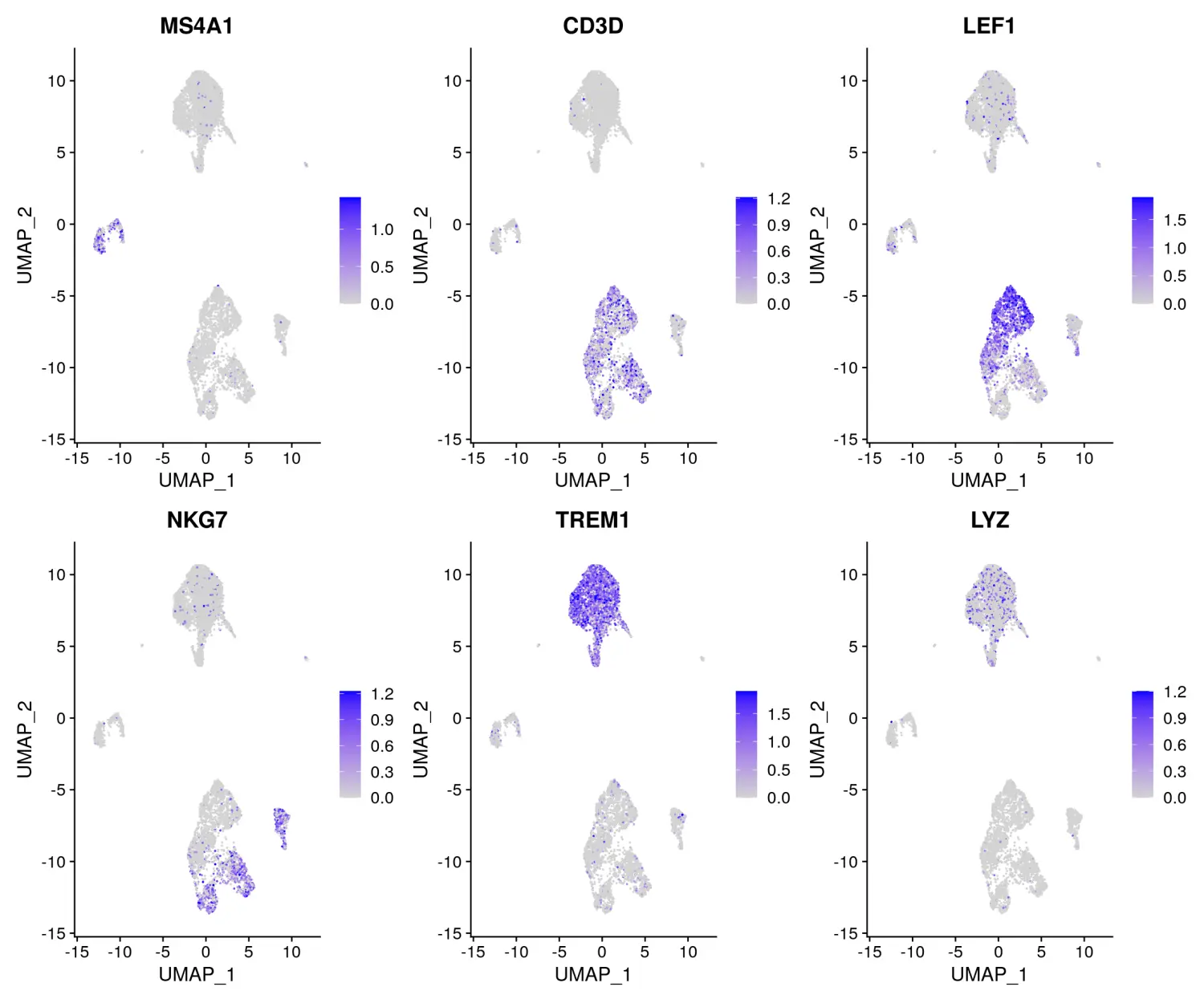
与scRNA-seq数据进行整合
为了帮助解释scATAC-seq测序数据,我们还可以基于来自同一生物系统(人PBMC)的scRNA-seq实验数据对细胞进行分类。在这里,我们利用跨模式整合和标签传输的方法,将scATAC-seq数据和scRNA-seq数据进行整合。我们基于scATAC-seq数据的基因活性矩阵和scRNA-seq数据集的基因表达矩阵识别它们之间共享的相关模式,以鉴定两种模式下匹配的生物学状态。该过程根据每个scRNA-seq定义的簇标签返回每个细胞的分类评分。
# Load the pre-processed scRNA-seq data for PBMCs
# 加载预处理的PBMC的scRNA-seq数据
pbmc_rna <- readRDS("/home/dongwei/scRNA-seq/pbmc_10k_v3.rds")
# 使用FindTransferAnchors函数识别整合的anchors
transfer.anchors <- FindTransferAnchors(
reference = pbmc_rna,
query = pbmc,
reduction = 'cca'
)
# 使用TransferData函数基于识别出的anchors进行数据映射整合
predicted.labels <- TransferData(
anchorset = transfer.anchors,
refdata = pbmc_rna$celltype,
weight.reduction = pbmc[['lsi']],
dims = 2:30
)
pbmc <- AddMetaData(object = pbmc, metadata = predicted.labels)
# 数据可视化
plot1 <- DimPlot(
object = pbmc_rna,
group.by = 'celltype',
label = TRUE,
repel = TRUE) + NoLegend() + ggtitle('scRNA-seq')
plot2 <- DimPlot(
object = pbmc,
group.by = 'predicted.id',
label = TRUE,
repel = TRUE) + NoLegend() + ggtitle('scATAC-seq')
plot1 + plot2
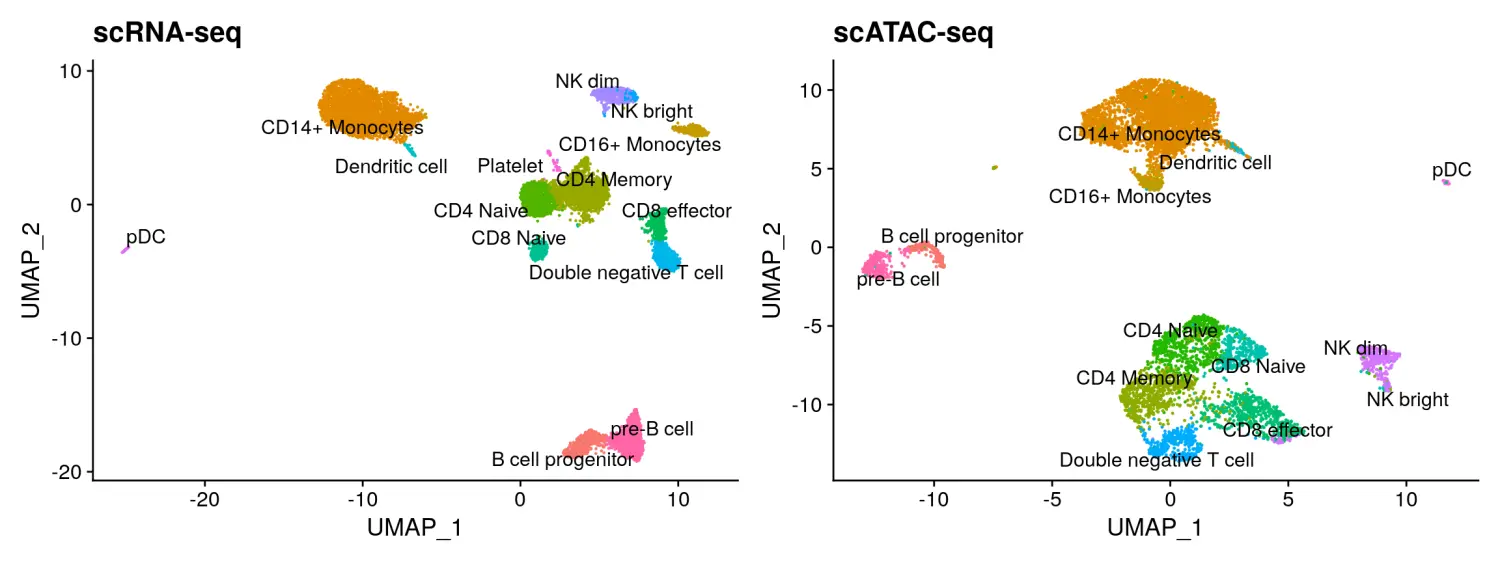
pbmc <- subset(pbmc,idents = 14, invert = TRUE)
pbmc <- RenameIdents(
object = pbmc,
'0' = 'CD14 Mono',
'1' = 'CD4 Memory',
'2' = 'CD8 Effector',
'3' = 'CD4 Naive',
'4' = 'CD14 Mono',
'5' = 'CD8 Naive',
'6' = 'DN T',
'7' = 'NK CD56Dim',
'8' = 'pre-B',
'9' = 'CD16 Mono',
'10' = 'pro-B',
'11' = 'DC',
'12' = 'NK CD56bright',
'13' = 'pDC'
)
鉴定不同聚类群之间差异可及性peaks区域
为了鉴定出不同细胞簇之间的差异可及性区域,我们可以进行差异可及性(DA)检验分析,并使用不同的函数进行可视化展示。正如Ntranos等人所建议的,我们使用逻辑回归模型(LR)进行DA分析。
# switch back to working with peaks instead of gene activities
DefaultAssay(pbmc) <- 'peaks'
# 使用FindMarkers函数进行差异分析
da_peaks <- FindMarkers(
object = pbmc,
ident.1 = "CD4 Naive",
ident.2 = "CD14 Mono",
min.pct = 0.2,
test.use = 'LR',
latent.vars = 'peak_region_fragments'
)
# 查看差异分析的结果
head(da_peaks)
## p_val avg_logFC pct.1 pct.2 p_val_adj
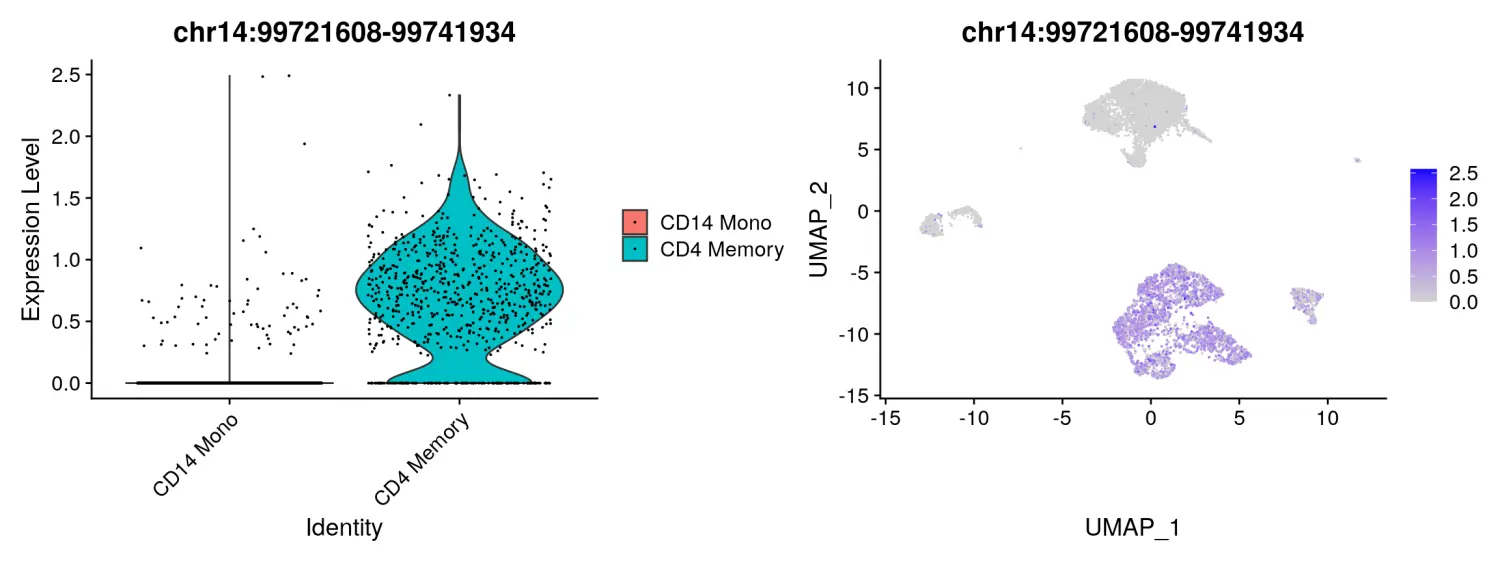
## chr14:99721608-99741934 0.000000e+00 0.7794737 0.847 0.024 0.000000e+00
## chr7:142501666-142511108 2.470763e-273 0.7166266 0.750 0.030 2.206565e-268
## chr17:80084198-80086094 1.971072e-263 1.1182759 0.666 0.007 1.760306e-258
## chr14:99695477-99720910 1.285851e-261 0.7604066 0.781 0.023 1.148355e-256
## chr2:113581628-113594911 3.004971e-238 -0.8896770 0.050 0.694 2.683649e-233
## chr6:44025105-44028184 2.263553e-229 -0.8723221 0.056 0.657 2.021511e-224
# 差异分析结果可视化
plot1 <- VlnPlot(
object = pbmc,
features = rownames(da_peaks)[1],
pt.size = 0.1,
idents = c("CD4 Memory","CD14 Mono")
)
plot2 <- FeaturePlot(
object = pbmc,
features = rownames(da_peaks)[1],
pt.size = 0.1
)
plot1 | plot2
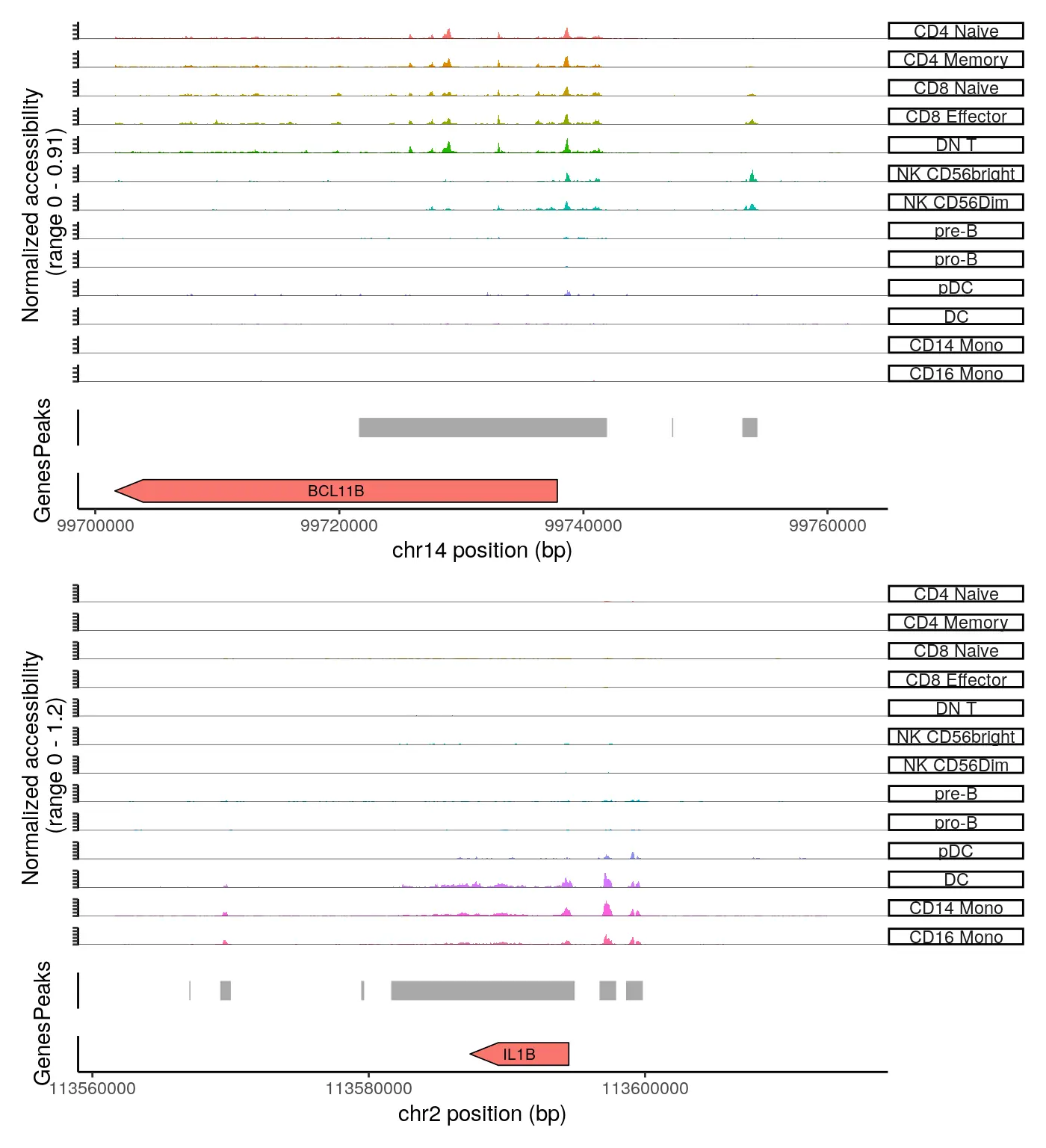
我们还可以使用ClosestFeature函数提供EnsDb中基因的注释信息,找到与每个peak最接近的基因。如果我们查看基因列表,会发现naive T细胞中的开放峰与BCL11B和GATA3(T细胞分化的关键调控因子)等基因接近,而单核细胞中开放的峰与CEBPB(单核细胞分化的关键调控因子)基因接近。我们还可以对ClosestFeature返回的基因集进行GO富集分析。
# 提取显著的差异可及性peak区域信息
open_cd4naive <- rownames(da_peaks[da_peaks$avg_logFC > 0.5, ])
open_cd14mono <- rownames(da_peaks[da_peaks$avg_logFC < -0.5, ])
# 使用ClosestFeature函数对差异开放性peak区域进行注释
closest_genes_cd4naive <- ClosestFeature(
regions = open_cd4naive,
annotation = gene.ranges,
sep = c(':', '-')
)
closest_genes_cd14mono <- ClosestFeature(
regions = open_cd14mono,
annotation = gene.ranges,
sep = c(':', '-')
)
# 查看注释后的结果
head(closest_genes_cd4naive)
## gene_id gene_name gene_biotype seq_coord_system
## ENSG00000127152 ENSG00000127152 BCL11B protein_coding chromosome
## ENSG00000204983 ENSG00000204983 PRSS1 protein_coding chromosome
## ENSG00000176155 ENSG00000176155 CCDC57 protein_coding chromosome
## ENSG00000127152.1 ENSG00000127152 BCL11B protein_coding chromosome
## ENSG00000134709 ENSG00000134709 HOOK1 protein_coding chromosome
## ENSG00000134954 ENSG00000134954 ETS1 protein_coding chromosome
## symbol entrezid closest_region
## ENSG00000127152 BCL11B 64919 chr14:99635624-99737861
## ENSG00000204983 PRSS1 5645, 5644 chr7:142457319-142460923
## ENSG00000176155 CCDC57 284001 chr17:80059336-80170706
## ENSG00000127152.1 BCL11B 64919 chr14:99635624-99737861
## ENSG00000134709 HOOK1 51361 chr1:60280458-60342050
## ENSG00000134954 ETS1 2113 chr11:128328656-128457453
## query_region distance
## ENSG00000127152 chr14:99721608-99741934 0
## ENSG00000204983 chr7:142501666-142511108 40742
## ENSG00000176155 chr17:80084198-80086094 0
## ENSG00000127152.1 chr14:99695477-99720910 0
## ENSG00000134709 chr1:60279767-60281364 0
## ENSG00000134954 chr11:128334097-128348572 0
head(closest_genes_cd14mono)
## gene_id gene_name gene_biotype seq_coord_system
## ENSG00000125538 ENSG00000125538 IL1B protein_coding chromosome
## ENSG00000181577 ENSG00000181577 C6orf223 protein_coding chromosome
## ENSG00000172216 ENSG00000172216 CEBPB protein_coding chromosome
## ENSG00000138621 ENSG00000138621 PPCDC protein_coding chromosome
## ENSG00000186350 ENSG00000186350 RXRA protein_coding chromosome
## ENSG00000273269 ENSG00000273269 RP11-761B3.1 protein_coding chromosome
## symbol entrezid closest_region
## ENSG00000125538 IL1B 3553 chr2:113587328-113594480
## ENSG00000181577 C6orf223 221416 chr6:43968317-43973695
## ENSG00000172216 CEBPB 1051 chr20:48807376-48809212
## ENSG00000138621 PPCDC 60490 chr15:75315896-75409803
## ENSG00000186350 RXRA 6256 chr9:137208944-137332431
## ENSG00000273269 RP11-761B3.1 NA chr2:47293080-47403650
## query_region distance
## ENSG00000125538 chr2:113581628-113594911 0
## ENSG00000181577 chr6:44025105-44028184 51409
## ENSG00000172216 chr20:48889794-48893313 80581
## ENSG00000138621 chr15:75334903-75336779 0
## ENSG00000186350 chr9:137263243-137268534 0
## ENSG00000273269 chr2:47297968-47301173 0
我们还可以使用CoveragePlot函数对按聚类分组的任何基因组区域绘制覆盖图进行可视化展示。These represent ‘pseudo-bulk’ accessibility tracks, where all cells within a cluster have been averaged together, in order to visualize a more robust chromatin landscape (thanks to Andrew Hill for giving the inspiration for this function in his excellent blog post).
# set plotting order
levels(pbmc) <- c("CD4 Naive","CD4 Memory","CD8 Naive","CD8 Effector","DN T","NK CD56bright","NK CD56Dim","pre-B",'pro-B',"pDC","DC","CD14 Mono",'CD16 Mono')
# 使用CoveragePlot函数绘制peaks的覆盖图
CoveragePlot(
object = pbmc,
region = rownames(da_peaks)[c(1,5)],
sep = c(":", "-"),
peaks = StringToGRanges(rownames(pbmc), sep = c(":", "-")),
annotation = gene.ranges,
extend.upstream = 20000,
extend.downstream = 20000,
ncol = 1
)
Working with datasets that were not quantified using CellRanger
10x Genomics的CellRanger软件可以为每个细胞生成几个有用的QC指标,以及峰/细胞矩阵和索引片段文件。在上面的示例中,我们直接使用了CellRanger的输出结果。但是,对于不是其产生的结果,Signac中也提供了许多类似的替代功能。
Generating a peak/cell or bin/cell matrix
我们可以使用FeatureMatrix函数生成一个计数矩阵。
# not run
# peak_ranges should be a set of genomic ranges spanning the set of peaks to be quantified per cell
peak_matrix <- FeatureMatrix(
fragments = fragment.path,
features = peak_ranges
)
为了方便起见,我们还提供了GenomeBinMatrix函数,该函数将生成一个跨越整个基因组的一组基因组范围,并在内部运行FeatureMatrix以生成基因组bin/细胞矩阵
# not run
bin_matrix <- GenomeBinMatrix(
fragments = fragment.path,
genome = 'hg19',
binsize = 5000
)
Counting fraction of reads in peaks
对于一个给定的峰/细胞或bin/细胞矩阵,我们可以使用FRiP函数计算每个细胞中peaks比对到的reads的比例。
# not run
pbmc <- FRiP(
object = pbmc,
peak.assay = 'peaks',
bin.assay = 'bins'
)
Counting fragments in genome blacklist regions
我们知道,基因组blacklist区域内比对到的reads的比率,可以作为低质量细胞预测的指标。Signac包中提供了几个参考基因组(hg19,hg38,mm9,mm10,ce10,ce11,dm3,dm6)的blacklist区域坐标,这些区域信息由ENCODE联盟提供。我们可以使用FractionCountsInRegion函数计算每个细胞在给定区域集中的比对到的reads的分数。
# not run
pbmc$blacklist_fraction <- FractionCountsInRegion(
object = pbmc,
assay = 'peaks',
regions = blacklist_hg19
)
参考来源:https://satijalab.org/signac/articles/pbmc_vignette.html

若有收获,就点个赞吧
0 人点赞
