SingleR包简介
SingleR是一个用于对单细胞RNA-seq测序(scRNA-seq)数据进行细胞类型自动注释的R包(Aran et al.2019)。它通过给定的具有已知类型标签的细胞样本作为参考数据集,对测试数据集中与参考集相似的细胞进行标记注释。具体来说,对于每个测试细胞:
- 首先,我们计算每个细胞的表达谱与参考样品的表达谱之间的Spearman相关性。这是通过在所有标记对之间识别的marker基因的并集完成的。
- 其次,我们将每个标签的分数定义为相关分布的固定分位数(默认为0.8)。
- 最后,我们对所有的标签重复此操作,然后将得分最高的标签作为此细胞的注释。
同时,该程序包还提供了一个网页版的工具,可以在线对单细胞转录组数据进行细胞类型注释分析,可以通过以下网址进行访问: https://comphealth.ucsf.edu/app/singler
参考文献:
Aran, Looney, Liu et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nature Immunology (2019)
SingleR包的安装
使用devtools包进行安装,如果安装失败,可下载进行本地安装,需提前安装好报错的各种依赖包。
# 使用devtools包进行安装devtools::install_github('dviraran/SingleR')# this might take long, though mostly because of the installation of Seurat.# 下载本地安装# 安装依赖包install.packages(c("outliers", "pbmcapply", "doFuture"))BiocManager::install(c("GSVA","singscore"))# 安装SingleR包install.packages("~/SingleR.tar.gz", repos = NULL, type = "source")
Using the built-in references
SingleR包提供了多个参考数据集(主要来自bulk RNA-seq or microarray数据)。例如,我们可以使用HumanPrimaryCellAtlasData()函数从Human Primary Cell Atlas中获取参考数据,该函数返回一个SummarizedExperiment对象,该对象中包含带有样本级标签的对数表达值矩阵。
# 加载SingleR包library(SingleR)# 使用HumanPrimaryCellAtlasData()函数加载参考数据集hpca.se <- HumanPrimaryCellAtlasData()# 查看参考数据集hpca.se## class: SummarizedExperiment## dim: 19363 713## metadata(0):## assays(1): logcounts## rownames(19363): A1BG A1BG-AS1 ... ZZEF1 ZZZ3## rowData names(0):## colnames(713): GSM112490 GSM112491 ... GSM92233 GSM92234## colData names(3): label.main label.fine label.ont
这里,我们使用La Manno et al.(2016)文章的数据作为测试数据集。为了加快分析的速度,我们仅使用前100个细胞的数据。
library(scRNAseq)# 加载测试数据集hESCs <- LaMannoBrainData('human-es')# 选取前100个细胞hESCs <- hESCs[,1:100]# SingleR() expects log-counts, but the function will also happily take# raw counts for the test dataset. The reference, however, must have log-values.library(scater)# 进行log归一化处理hESCs <- logNormCounts(hESCs)
我们使用hpca.se数据作为参考数据集,通过SingleR()函数对hESCs测试数据集中的每个细胞进行类型注释。注意,默认的标记检测方法是采用每个基因的每个标签中位数的正对数倍变化最大的基因。
# 使用SingleR函数进行细胞类型注释pred.hesc <- SingleR(test = hESCs, ref = hpca.se, labels = hpca.se$label.main)# 查看细胞注释结果pred.hesc## DataFrame with 100 rows and 5 columns## scores first.labels## <matrix> <character>## 1772122_301_C02 0.347652:0.109547:0.123901:... Neuroepithelial_cell## 1772122_180_E05 0.361187:0.134934:0.148672:... Neuroepithelial_cell## 1772122_300_H02 0.446411:0.190084:0.222594:... Neuroepithelial_cell## 1772122_180_B09 0.373512:0.143537:0.164743:... Neuroepithelial_cell## 1772122_180_G04 0.357341:0.126511:0.141987:... Neuroepithelial_cell## ... ... ...## 1772122_299_E07 0.371989:0.169379:0.1986877:... Neuroepithelial_cell## 1772122_180_D02 0.353314:0.115864:0.1374981:... Neuroepithelial_cell## 1772122_300_D09 0.348789:0.136732:0.1303042:... Neuroepithelial_cell## 1772122_298_F09 0.332361:0.141439:0.1437860:... Neuroepithelial_cell## 1772122_302_A11 0.324928:0.101609:0.0949826:... Neuroepithelial_cell## tuning.scores labels pruned.labels## <DataFrame> <character> <character>## 1772122_301_C02 0.1824402:0.0991116 Neuroepithelial_cell Neuroepithelial_cell## 1772122_180_E05 0.1375484:0.0647134 Neurons Neurons## 1772122_300_H02 0.2757982:0.1369690 Neuroepithelial_cell Neuroepithelial_cell## 1772122_180_B09 0.0851623:0.0819878 Neuroepithelial_cell Neuroepithelial_cell## 1772122_180_G04 0.1988415:0.1016622 Neuroepithelial_cell Neuroepithelial_cell## ... ... ... ...## 1772122_299_E07 0.1760025:0.0922504 Neuroepithelial_cell Neuroepithelial_cell## 1772122_180_D02 0.1967609:0.1124805 Neuroepithelial_cell Neuroepithelial_cell## 1772122_300_D09 0.0816424:0.0221368 Neuroepithelial_cell Neuroepithelial_cell## 1772122_298_F09 0.1872499:0.0671893 Neuroepithelial_cell Neuroepithelial_cell## 1772122_302_A11 0.1560800:0.1051322 Astrocyte Astrocyte
在输出的DataFrame中,每一行包含了每个细胞注释的预测结果。每一列显示了标签在微调之前(first.labels),微调之后(labels)和修剪后(pruned.labels)以及相关得分。
table(pred.hesc$labels)#### Astrocyte Neuroepithelial_cell Neurons## 14 81 5
Using single-cell references
这里,我们使用来自scRNAseq包中的两个人类胰腺数据集。目的是使用一个预先标记好的数据集对另一个未标记的数据集进行细胞类型注释。首先,我们使用Muraro et al.(2016)的数据作为我们的参考数据集。
# 加载scRNAseq包library(scRNAseq)# 加载参考数据集sceM <- MuraroPancreasData()# One should normally do cell-based quality control at this point, but for# brevity's sake, we will just remove the unlabelled libraries here.# 删除未标记的细胞sceM <- sceM[,!is.na(sceM$label)]# 对表达数据进行log归一化处理sceM <- logNormCounts(sceM)
接下来,我们使用Grun et al.(2016)的数据作为测试数据集。为了加快分析的速度,我们挑选前100个细胞进行分析。
# 加载测试数据集sceG <- GrunPancreasData()sceG <- sceG[,colSums(counts(sceG)) > 0] # Remove libraries with no counts.sceG <- logNormCounts(sceG)sceG <- sceG[,1:100]
然后,我们使用SingleR()函数进行细胞类型注释分析,但要使用标记检测模式(marker detection mode),该模式会考虑跨细胞表达的差异。在这里,我们将使用Wilcoxon ranked sum test检验来识别标签对之间比较的top markers。与默认的标记检测算法相比,此方法会更慢,但更适用于单细胞测序数据(对于中位数通常为零的低覆盖率数据,它可能会失败)。
# 使用SingleR函数进行细胞类型注释,并指定de.method="wilcox"检测方法pred.grun <- SingleR(test=sceG, ref=sceM, labels=sceM$label, de.method="wilcox")# 查看细胞类型注释的预测结果table(pred.grun$labels)#### acinar beta delta duct## 53 4 2 41
Annotation diagnostics
Based on the scores within cells
SingleR包提供了一些常用的可视化工具。我们可以使用plotScoreHeatmap()函数显示所有参考标签中所有细胞的分数,这可以让我们检查整个数据集中预测标签的置信度。每个细胞的实际分配标签显示在顶部的颜色栏中。注意,如果进行了微调,这可能不是视觉上得分最高的标签,因为只有预先调整的分数才能在所有标签上直接比较。
plotScoreHeatmap(pred.grun)
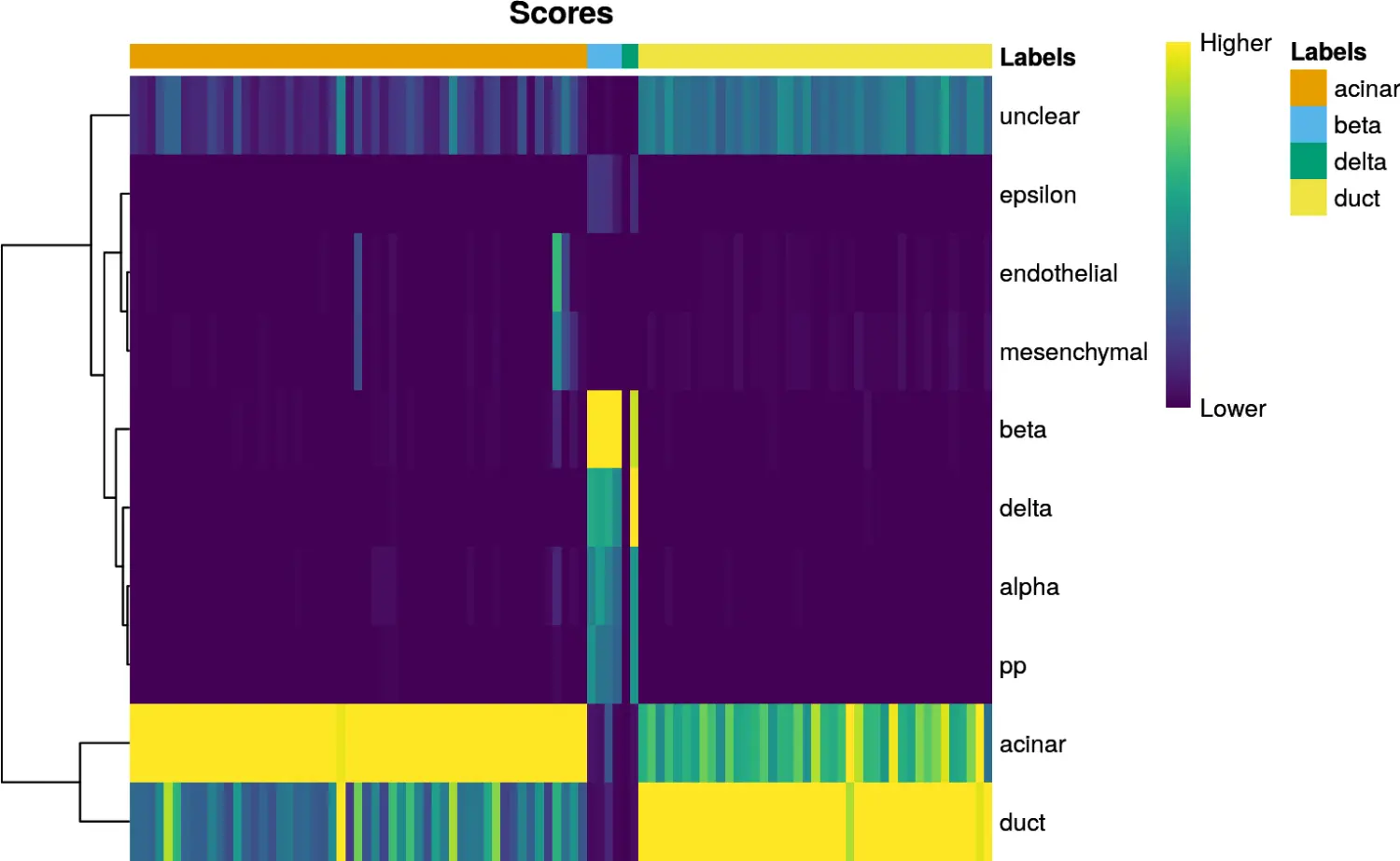
在上图中,我们主要是检查每个细胞内得分的分布。理想情况下,每个细胞(即热图的列)应具有一个明显比其他细胞高的分数,这表明它已被明确地分配给单个标签。
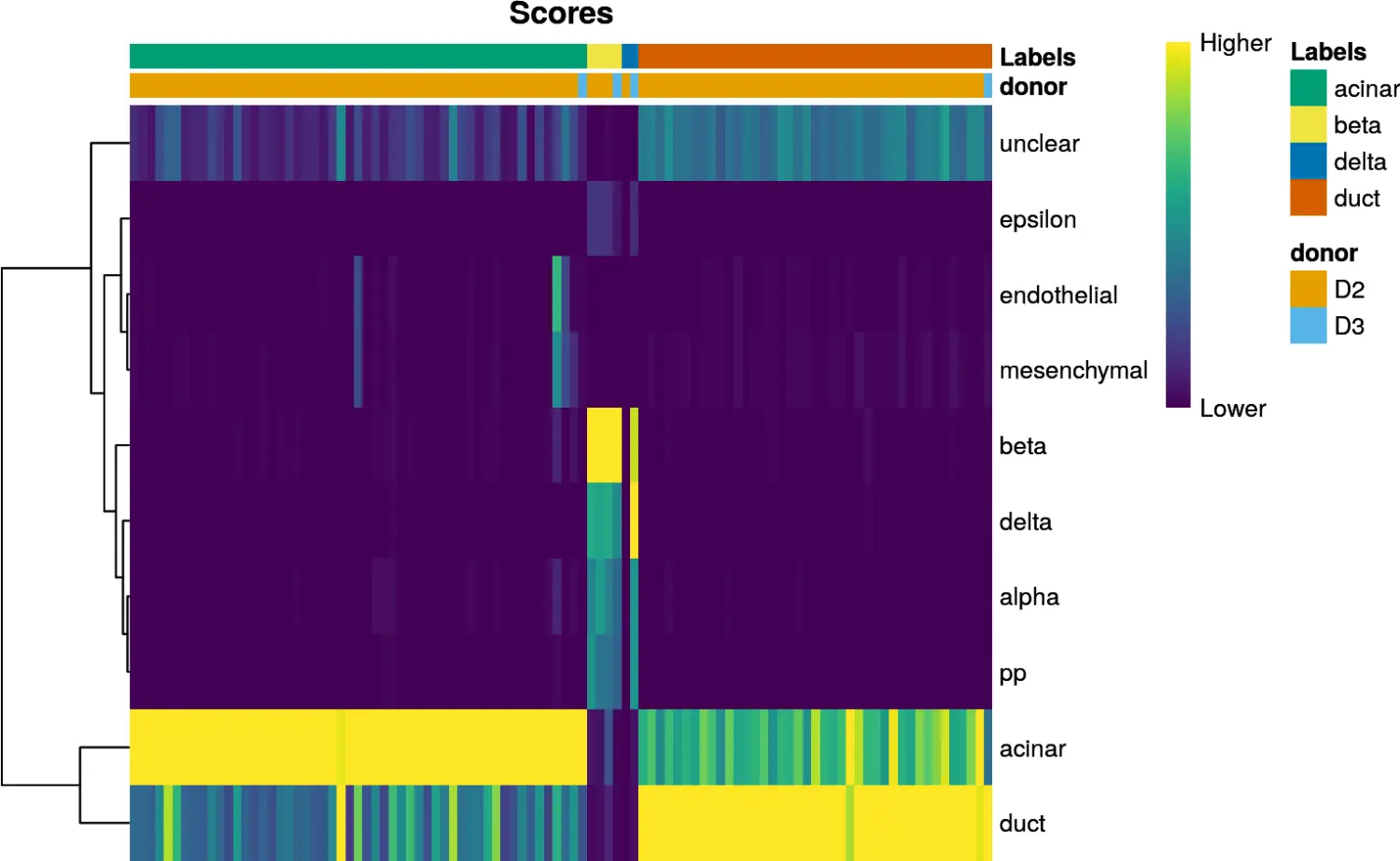
我们还可以通过设置clusters=或annotation_col=参数来显示每个细胞的其他元数据信息。通常,这对于检查潜在的批次效应,不同条件之间的细胞类型组成差异,无监督聚类中clusters之间的关系等很有用。在下面的代码中,我们显示每个细胞来自哪个供体:
plotScoreHeatmap(pred.grun,annotation_col=as.data.frame(colData(sceG)[,"donor",drop=FALSE]))
Based on the deltas across cells
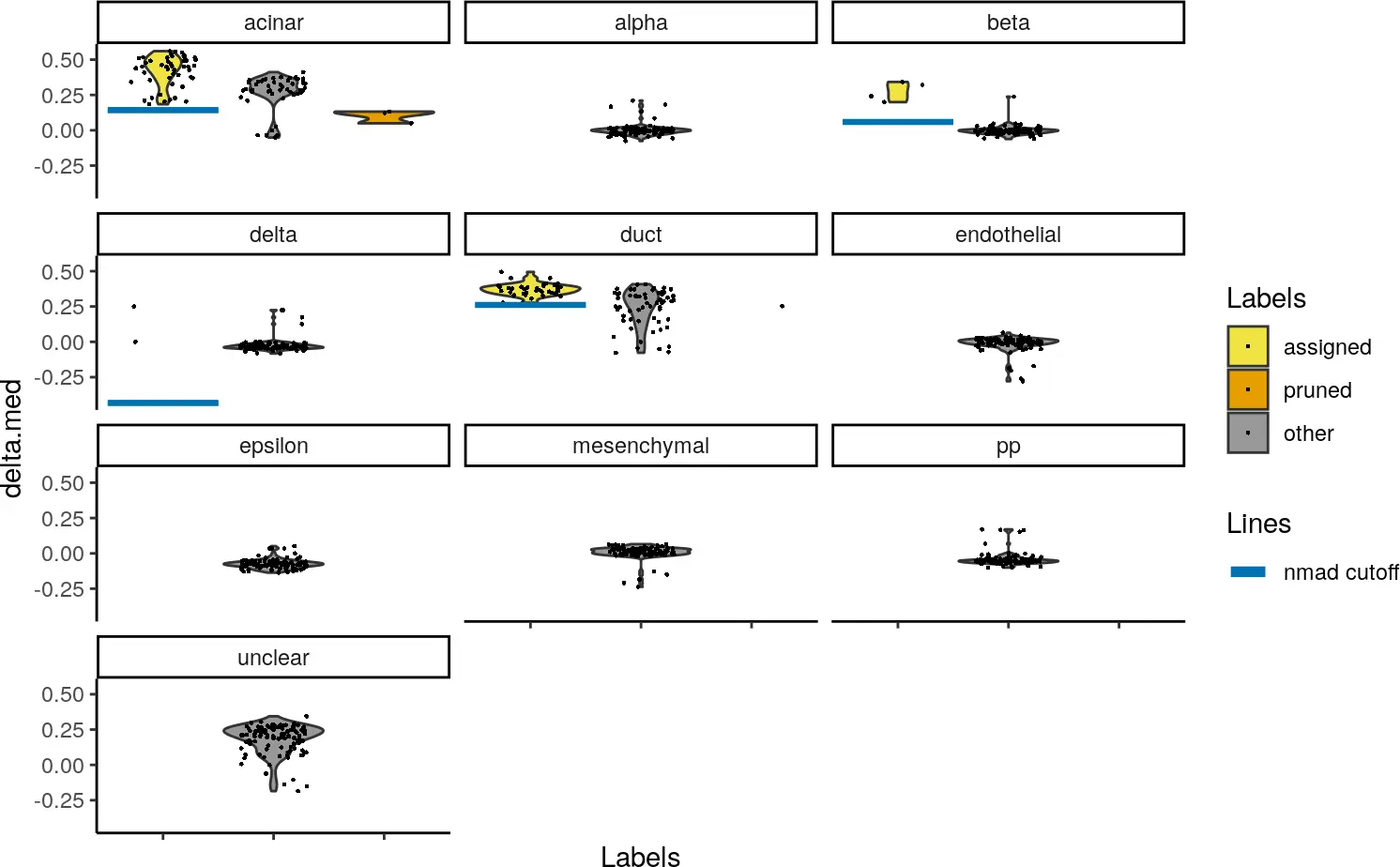
我们还可以使用pruneScores()函数删除那些质量低下或模棱两可的分配。具体来说,我们可以基于每个细胞的“delta”值(即,所分配的标签的得分与每个细胞的所有标签的中位数之间的差值)来识别分配不明确的细胞。低delta值的标记表示分配是不确定的。pruneScores()函数使用基于异常值的方法来确定用于修剪的确切阈值,该方法考虑了各种情况下相关程度的差异。
to.remove <- pruneScores(pred.grun)summary(to.remove)## Mode FALSE TRUE## logical 96 4
默认情况下,SingleR()函数会以pruned.labels字段来报告出修剪的标签,其中低质量的分配以NA表示。但是,默认的修剪阈值可能不适用于每个数据集。我们提供了plotScoreDistribution()函数来通过使用具有相同标签的细胞中的信息来帮助确定阈值是否合适。这将显示每个标记的跨细胞分布,其中pruneScores()函数以低于3个中位数绝对偏差(MAD)值来定义适当的阈值。
plotScoreDistribution(pred.grun, show = "delta.med", ncol = 3, show.nmads = 3)
如果必须调整某些参数,我们可以直接使用调整后的参数来调用pruneScores()函数。这里,我们将要丢弃的标签设置为NA,这也是SingleR()函数如何在pruned.labels中标记此类标签的方式。
new.pruned <- pred.grun$labelsnew.pruned[pruneScores(pred.grun, nmads=5)] <- NAtable(new.pruned, useNA="always")## new.pruned## acinar beta delta duct <NA>## 53 4 2 41 0
Based on marker gene expression
另一个简单而有效的诊断方法是检查测试数据集中每个标记的marker基因的表达。我们从SingleR()结果的元数据中提取标记的标识,并使用scater包中的plotHeatmap()函数进行可视化展示。如果测试数据集中的某个细胞确信地分配给了特定标签,我们希望标记为该标签的marker基因具有很高的表达。
all.markers <- metadata(pred.grun)$de.genessceG$labels <- pred.grun$labels# Beta cell-related markersplotHeatmap(sceG, order_columns_by="labels",features=unique(unlist(all.markers$beta)))
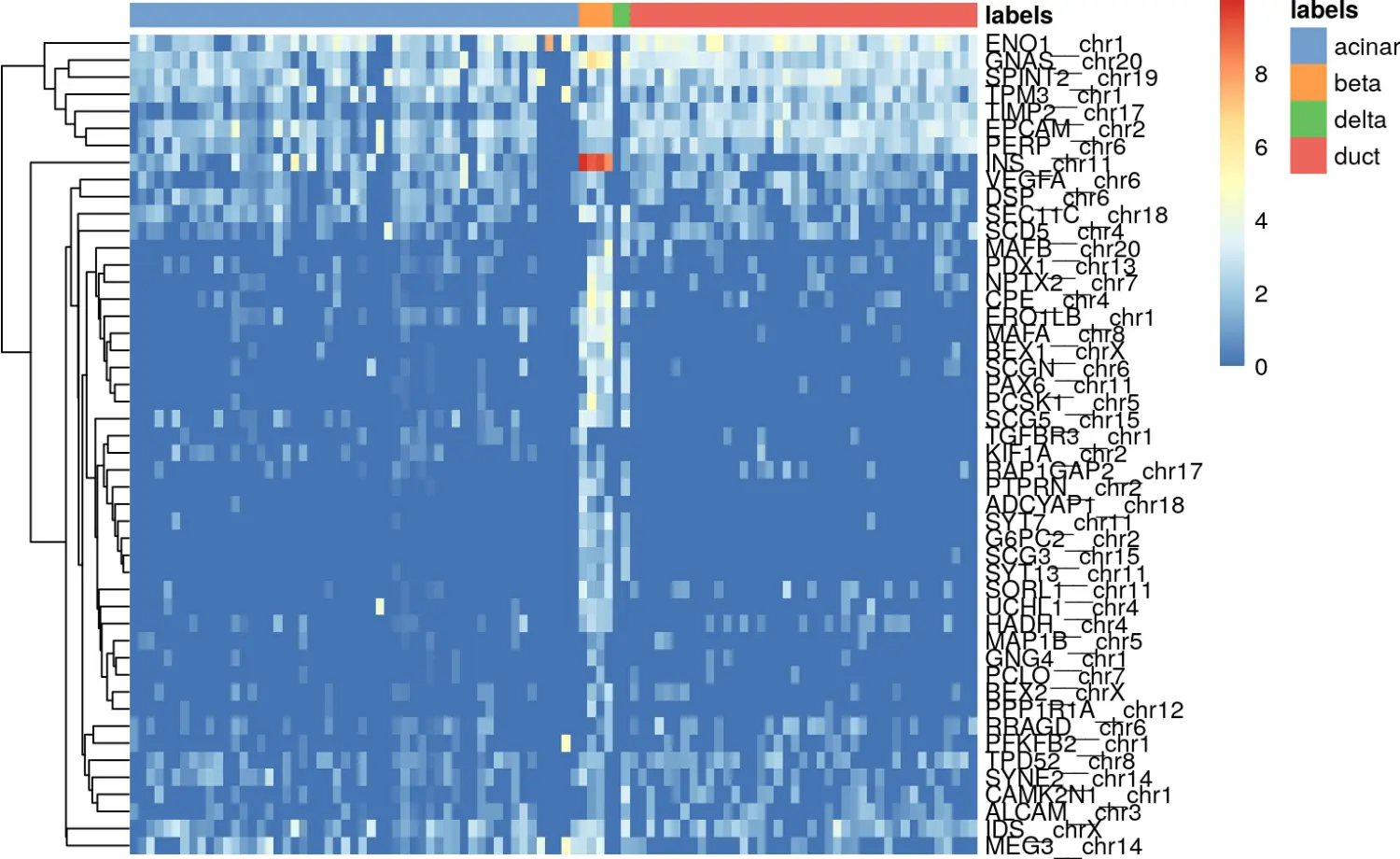
We can similarly perform this for all labels by wrapping this code in a loop, as shown below:
for (lab in unique(pred.grun$labels)) {plotHeatmap(sceG, order_columns_by=list(I(pred.grun$labels)),features=unique(unlist(all.markers[[lab]])))}
Available references
旧版的SingleR包中提供了RDA文件,该文件中包含了来源于以下bulk RNA-seq, microarray和single-cell RNA-seq测序的标准化后的表达值和细胞类型标签:
- Blueprint (Martens and Stunnenberg 2013) and Encode (The ENCODE Project Consortium 2012),
- the Human Primary Cell Atlas (Mabbott et al. 2013),
- the murine ImmGen (Heng et al. 2008), and
- a collection of mouse data sets downloaded from GEO (Benayoun et al. 2019).
前三个参考数据集的bulk RNA-seq和microarray数据是从预先分选好的细胞群体中获得的,即这些样品中的细胞标记大多是根据各自的分类/纯化策略而不是通过计算机模拟得出的方法。
我们还从免疫细胞的bulk RNA-seq和microarray数据中准备了三个附加的参考数据集。这些数据集中的每一个细胞也都是从预先分类好的的细胞群体中获得的:
- The Database for Immune Cell Expression(/eQTLs/Epigenomics) (Schmiedel et al. 2018),
- Novershtern Hematopoietic Cell Data - GSE24759 - formerly known as Differentiation Map (Novershtern et al. 2011), and
- Monaco Immune Cell Data - GSE107011 (Monaco et al. 2019).
The characteristics of each dataset are summarized below: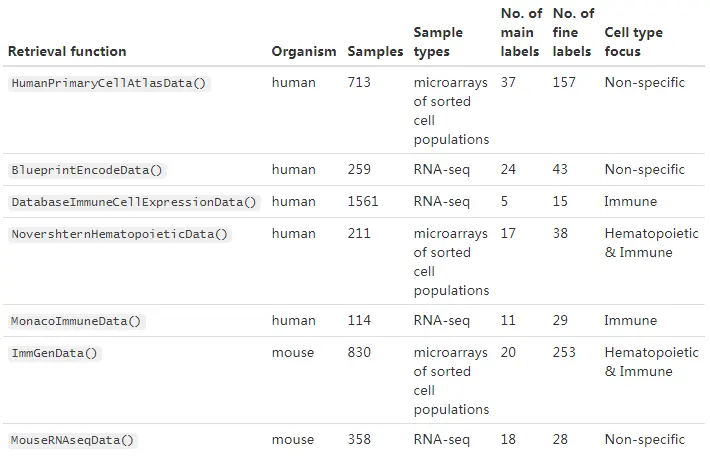
提供的参考数据集标签:
Reference options
Pseudo-bulk aggregation
Single-cell reference参考数据集提供了与我们的测试数据集进行相似性比较,从而希望对测试数据集进行更准确的分类注释。但是,与bulk references参考相比,single-cell references参考中经常有更多的样本,从而增加了对细胞分类注释的计算工作。我们可以将所有的细胞聚合汇总为一个“pseudo-bulk”样本(例如,通过对所有细胞表达的log值取平均值)并将其用作参考,这使我们获得了与使用bulk references相同的效率。
使用这种方法的代价是,我们丢弃了有关每个标签内细胞分布的潜在有用信息。如果属于异质细胞群体中的细胞远离了群体中心,则可能无法正确的分配它们。我们试图通过在每个细胞内使用k-均值聚类来创建代表特定空间区域的pseudo-bulk样本来保留一些此类信息。
这种聚合方法是通过aggregateReferences()函数来实现的,下面对Muraro et al.(2016)的数据进行演示。该函数返回一个SummarizedExperiment对象,其中包含pseudo-bulk的表达信息和相应的标签。
set.seed(100) # for the k-means step.# 使用aggregateReference函数对参考数据集进行聚合aggr <- aggregateReference(sceM, labels=sceM$label)aggr## class: SummarizedExperiment## dim: 19059 116## metadata(0):## assays(1): logcounts## rownames(19059): A1BG-AS1__chr19 A1BG__chr19 ... ZZEF1__chr17## ZZZ3__chr1## rowData names(0):## colnames(116): alpha.1 alpha.2 ... mesenchymal.8 epsilon.1## colData names(1): label
The resulting SummarizedExperiment can then be used as a reference in SingleR().
# 使用SingleR函数进行细胞类型注释pred.aggr <- SingleR(sceG, aggr, labels=aggr$label)table(pred.aggr$labels)#### acinar beta delta duct## 53 4 1 42
Using multiple references
在某些情况下,我们可能希望使用多个参考数据集来对测试数据进行标记注释。这样可以避免单个参考数据集中非覆盖到的标记,从而得到一组更加全面的细胞类型标记,尤其是在考虑分辨率差异的情况下。 我们可以通过将多个对象简单地传递到SingleR()函数中的ref=和label=参数,即可支持使用多个参考数据集。
# 加载另一个参考数据集Rbp.se <- BlueprintEncodeData()# 使用多个参考数据集进行细胞类型注释pred.combined <- SingleR(test = hESCs,ref = list(BP=bp.se, HPCA=hpca.se),labels = list(bp.se$label.main, hpca.se$label.main))
The output is the same form as previously described, and we can easily gain access to the combined set of labels:
table(pred.combined$labels)#### Astrocyte Neuroepithelial_cell Neurons## 6 64 30
我们的策略是,首先基于每个参考数据集分别进行细胞类型注释,然后在不同参考数据集中使用得分最高的标签。我们提供了一种轻量级的方法来合并多个参考数据集的信息,同时还避免了批处理效应和前期协调的需要。(当然,这种方法的主要困难是同一细胞类型在不同参考数据集中可能具有不同的标签,这将在解释过程中需要进行一些隐式的协调)。
Harmonizing labels
我们还提供了matchReferences()函数,可以为两个参考数据集之间的标签协调提供一种简单而优雅的方法。每个参考数据集会进行相互注释,并计算每对标签之间相互分配的概率。接近1的概率表示那对标签之间存在1:1的关系;另一方面,全零概率表示该标签相对于特定参考数据集是唯一的。
matched <- matchReferences(bp.se, hpca.se,bp.se$label.main, hpca.se$label.main)pheatmap::pheatmap(matched, col=viridis::plasma(100))
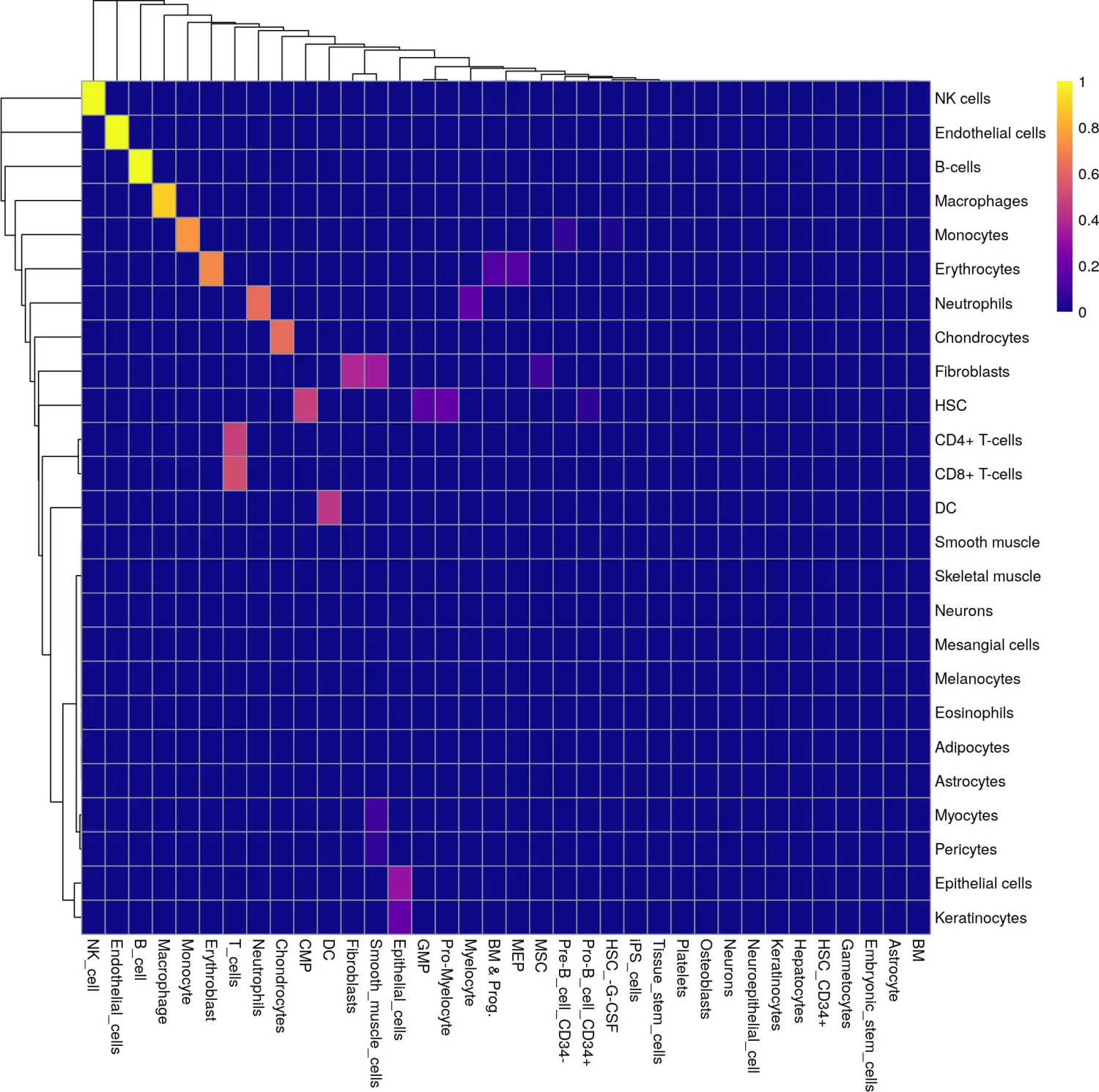
上面的热图可以用于指导我们对多个不同的数据集进行协调,使得在代表相同细胞类型或状态的所有标签上强制使用一致的词汇表。协调统一后最明显的好处是简化了结果的解释。但是,一个更为重要的好处是,使用协调统一的标签可以使分类机制防止多个参考数据集之间出现无关紧要的批次影响。例如,在使用SingleR()函数时,如果marker基因在多个参考数据集中一致性上调,那么它们将受到青睐,从而提高了测试数据集对技术特质的鲁棒性。
考虑到生物系统和技术差异带来的风险,我们强调在此过程中仍需要进行一些手动的调整干预。例如,在每个参考数据集中,神经元细胞被认为是唯一的,而HPCA数据集中的平滑肌细胞与Blueprint/ENCODE数据集中的成纤维细胞是不正确匹配的。CD4+和CD8+ T细胞也都分配给了“T细胞”,因此在这里我们需要对协调标记的可接受分辨率做出一些决定。
此外,我们还可以使用此功能来对两个独立的scRNA-seq数据之间鉴定相匹配的cluster簇。我们可以匹配聚类群并整合来自多个数据集的结果,从而避免了进行批次校正和重新聚类的困难。
Advanced use
Improving efficiency
我们还可以将SingleR()函数的工作流程分成两个独立的训练(training)和分类(classification)的步骤。这意味着只需要执行一次训练(例如,marker detection和assembling of nearest-neighbor indices)。然后,只要测试特征集与训练集中的特征相同或重叠,就可以在具有不同测试数据集的多个分类中重复使用所得的数据结构。例如:
common <- intersect(rownames(hESCs), rownames(hpca.se))# 使用trainSingleR函数构建训练集trained <- trainSingleR(hpca.se[common,], labels=hpca.se$label.main)# 使用classifySingleR函数基于训练集的结果进行细胞类型标记注释pred.hesc2 <- classifySingleR(hESCs[common,], trained)# 查看注释的结果table(pred.hesc$labels, pred.hesc2$labels)#### Astrocyte Neuroepithelial_cell Neurons## Astrocyte 14 0 0## Neuroepithelial_cell 0 81 0## Neurons 0 0 5
我们还可以使用以下参数来提高分析效率:
- 使用
BiocNeighbors包中的BNPARAM=参数设置trainSingleR()函数中的最近邻搜索的近似算法。 - 使用
BiocParallel包中的BPPARAM=参数并行化运行classifySingleR()函数中的微调步骤。
这些参数也可以在SingleR()函数中指定使用。
Defining custom markers
我们还可以使用任何DE检验的方法来构建自己的marker基因列表。例如,我们可以使用scran包中的pairwiseTTests方法进行配对的t检验,并从每个成对比较中获取top10的marker基因。
library(scran)# 使用pairwiseTTests函数进行差异分析out <- pairwiseTTests(logcounts(sceM), sceM$label, direction="up")# 使用getTopMarkers函数提取显著的top基因markers <- getTopMarkers(out$statistics, out$pairs, n=10)
然后,我们使用genes=参数将这些marker基因提供给SingleR()函数。与默认方法相比,更加focused的基因集可以更快地进行注释分析。
pred.grun2 <- SingleR(test=sceG, ref=sceM, labels=sceM$label, genes=markers)
# 查看注释的结果
table(pred.grun2$labels)
##
## acinar beta delta duct pp unclear
## 59 4 1 34 1 1
在某些情况下,一些markers基因可能仅可用于特定的标签,而不能用于标签之间的成对比较。我们可以通过向基因提供character vectors的命名列表来解决此问题。请注意,由于有关成对差异的信息已被丢弃,这可能不如list-of-lists方法强大。
label.markers <- lapply(markers, unlist, recursive=FALSE)
pred.grun3 <- SingleR(test=sceG, ref=sceM, labels=sceM$label, genes=label.markers)
table(pred.grun$labels, pred.grun3$labels)
##
## acinar beta delta duct pp
## acinar 51 0 0 2 0
## beta 0 4 0 0 0
## delta 0 0 1 0 1
## duct 2 0 0 39 0
参考来源: http://www.bioconductor.org/packages/release/bioc/vignettes/SingleR/inst/doc/SingleR.html

若有收获,就点个赞吧
0 人点赞
