输入/输出
Immunarch包提供了以下函数进行数据的读取和保存:
repLoad- to load the repertoires, having compatible format.repSave- to save changes and to write the repertoire data to a file in a specific format (immunarch, VDJtools).
其中,repLoad函数可以自动检测输入文件的格式,我们可以通过?repLoad查看更详细的数据导入信息。
目前,immunarch包可以支持以下免疫组库数据的格式:
"immunarch"- 当前的软件工具,以防您忘记它:)"immunoseq"- http://www.adaptivebiotech.com/immunoseq"mitcr"- https://github.com/milaboratory/mitcr"mixcr"- https://github.com/milaboratory/mixcr"migec"- http://migec.readthedocs.io/en/latest/"migmap"- https://github.com/mikessh/migmap"tcr"- https://imminfo.github.io/tcr/"vdjtools"- https://vdjtools-doc.readthedocs.io/en/master/"imgt"- http://www.imgt.org/HighV-QUEST/"airr"- http://docs.airr-community.org/en/latest/datarep/overview.html"10x"- https://support.10xgenomics.com/single-cell-vdj/software/pipelines/latest/output/annotation"archer"- ArcherDX clonotype tables. https://archerdx.com/immunology/
以下数据格式后续也会添加到该包中。
"imseq"- http://www.imtools.org/"rtcr"- https://github.com/uubram/RTCR"vidjil"- http://www.vidjil.org/
使用dplyr和 immunarch进行基本数据操作
获取丰度最高的克隆型
该函数返回给定TCR/BCR库中最丰富的克隆型:
top(immdata$data[[1]])## # A tibble: 10 x 15## Clones Proportion CDR3.nt CDR3.aa V.name D.name J.name V.end D.start D.end## <dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <int> <int> <int>## 1 173 0.0204 TGCGCC… CASSQE… TRBV4… TRBD1 TRBJ2… 16 18 26## 2 163 0.0192 TGCGCC… CASSYR… TRBV4… TRBD1 TRBJ2… 11 13 18## 3 66 0.00776 TGTGCC… CATSTN… TRBV15 TRBD1 TRBJ2… 11 16 22## 4 54 0.00635 TGTGCC… CATSIG… TRBV15 TRBD2 TRBJ2… 11 19 25## 5 48 0.00565 TGTGCC… CASSPW… TRBV27 TRBD1 TRBJ1… 11 16 23## 6 48 0.00565 TGCGCC… CASQGD… TRBV4… TRBD1 TRBJ1… 8 13 19## 7 40 0.00471 TGCGCC… CASSQD… TRBV4… TRBD1 TRBJ2… 16 21 26## 8 31 0.00365 TGTGCC… CASSEE… TRBV2 TRBD1 TRBJ1… 15 17 20## 9 30 0.00353 TGCGCC… CASSQP… TRBV4… TRBD1 TRBJ2… 14 23 28## 10 28 0.00329 TGTGCC… CASSWV… TRBV6… TRBD1 TRBJ2… 12 20 25## # … with 5 more variables: J.start <int>, VJ.ins <dbl>, VD.ins <dbl>,## # DJ.ins <dbl>, Sequence <lgl>
过滤functional/non-functional/in-frame/out-of-frame克隆型
方便的是,函数在数据框列表上被向量化;
使用coding(immdata$data)命令将返回编码序列的列表:
coding(immdata$data[[1]])# A tibble: 6,443 x 15# Clones Proportion CDR3.nt CDR3.aa V.name D.name J.name V.end D.start D.end J.start VJ.ins# <dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <int> <int> <int> <int> <dbl># 1 173 0.0204 TGCGCC… CASSQE… TRBV4… TRBD1 TRBJ2… 16 18 26 31 -1# 2 163 0.0192 TGCGCC… CASSYR… TRBV4… TRBD1 TRBJ2… 11 13 18 22 -1# 3 66 0.00776 TGTGCC… CATSTN… TRBV15 TRBD1 TRBJ2… 11 16 22 34 -1# 4 54 0.00635 TGTGCC… CATSIG… TRBV15 TRBD2 TRBJ2… 11 19 25 26 -1# 5 48 0.00565 TGTGCC… CASSPW… TRBV27 TRBD1 TRBJ1… 11 16 23 31 -1# 6 48 0.00565 TGCGCC… CASQGD… TRBV4… TRBD1 TRBJ1… 8 13 19 23 -1# 7 40 0.00471 TGCGCC… CASSQD… TRBV4… TRBD1 TRBJ2… 16 21 26 29 -1# 8 31 0.00365 TGTGCC… CASSEE… TRBV2 TRBD1 TRBJ1… 15 17 20 29 -1# 9 30 0.00353 TGCGCC… CASSQP… TRBV4… TRBD1 TRBJ2… 14 23 28 34 -1#10 28 0.00329 TGTGCC… CASSWV… TRBV6… TRBD1 TRBJ2… 12 20 25 28 -1# … with 6,433 more rows, and 3 more variables: VD.ins <dbl>, DJ.ins <dbl>, Sequence <lgl>
使用noncoding(immdata$data)命令返回非编码序列的列表:
noncoding(immdata$data[[1]])# A tibble: 89 x 15# Clones Proportion CDR3.nt CDR3.aa V.name D.name J.name V.end D.start D.end J.start VJ.ins# <dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <int> <int> <int> <int> <dbl># 1 12 0.00141 TGCGCC… CASSRP… TRBV2… TRBD1 TRBJ2… 12 17 24 27 -1# 2 2 0.000235 TGTGCC… CASSVA… TRBV7… TRBD2 TRBJ2… 11 15 23 29 -1# 3 2 0.000235 TGTGCC… CASSK*… TRBV2… TRBD2 TRBJ2… 14 19 23 29 -1# 4 2 0.000235 TGTGCC… CASSRG… TRBV28 TRBD2 TRBJ2… 10 12 19 24 -1# 5 1 0.000118 TGCAGT… CSALDG… TRBV2… TRBD2 TRBJ2… 7 18 23 26 -1# 6 1 0.000118 TGTGCC… CASSLD… TRBV7… TRBD1 TRBJ2… 15 16 24 27 -1# 7 1 0.000118 TGTGCC… CASSRT… TRBV6… TRBD1 TRBJ2… 11 13 20 28 -1# 8 1 0.000118 TGCGCC… CASSQV… TRBV4… TRBD2 TRBJ1… 15 21 27 34 -1# 9 1 0.000118 TGTGCC… CASSTD… TRBV2… TRBD2 TRBJ2… 12 15 25 27 -1# 10 1 0.000118 TGCAGC… CSE*QG… TRBV2… TRBD1 TRBJ1… 6 10 17 28 -1# … with 79 more rows, and 3 more variables: VD.ins <dbl>, DJ.ins <dbl>, Sequence <lgl>
Now, the computation of the number of filtered sequences is straightforward:
nrow(inframes(immdata$data[[1]]))# [1] 6445
And for the out-of-frame clonotypes:
nrow(outofframes(immdata$data[[1]]))# [1] 87
获取具有特定 V 基因的克隆型子集
根据指定索引中的标签对数据表进行子集化很简单。在示例中,结果数据框仅包含带有“TRBV10-1”V 基因的记录:
filter(immdata$data[[1]], V.name == 'TRBV10-1')## # A tibble: 24 x 15## Clones Proportion CDR3.nt CDR3.aa V.name D.name J.name V.end D.start D.end## <dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <int> <int> <int>## 1 2 0.000235 TGCGCC… CASSES… TRBV1… TRBD2 TRBJ2… 16 20 25## 2 2 0.000235 TGCGCC… CASSDG… TRBV1… TRBD1 TRBJ2… 13 15 22## 3 1 0.000118 TGCGCC… CASSGD… TRBV1… TRBD2 TRBJ2… 8 10 15## 4 1 0.000118 TGCGCC… CATLRS… TRBV1… TRBD1 TRBJ2… 6 7 9## 5 1 0.000118 TGCGCC… CASSES… TRBV1… TRBD2 TRBJ2… 16 20 22## 6 1 0.000118 TGCGCC… CASSES… TRBV1… TRBD2 TRBJ2… 16 17 21## 7 1 0.000118 TGCGCC… CASRAS… TRBV1… TRBD2 TRBJ2… 10 13 21## 8 1 0.000118 TGCGCC… CASRRD… TRBV1… TRBD1 TRBJ2… 8 13 19## 9 1 0.000118 TGCGCC… CASSEV… TRBV1… TRBD1 TRBJ2… 14 19 24## 10 1 0.000118 TGCGCC… CASSEG… TRBV1… TRBD2 TRBJ2… 13 19 27## # … with 14 more rows, and 5 more variables: J.start <int>, VJ.ins <dbl>,## # VD.ins <dbl>, DJ.ins <dbl>, Sequence <lgl>
Downsampling
# 使用repSample函数进行downsamplingds = repSample(immdata$data, "downsample", 100)sapply(ds, nrow)## A2-i129 A2-i131 A2-i133 A2-i132 A4-i191 A4-i192 MS1 MS2 MS3 MS4## 97 97 90 99 91 98 91 99 87 98## MS5 MS6## 88 100ds = repSample(immdata$data, "sample", .n = 10)sapply(ds, nrow)## A2-i129 A2-i131 A2-i133 A2-i132 A4-i191 A4-i192 MS1 MS2 MS3 MS4## 10 10 10 10 10 10 10 10 10 10## MS5 MS6## 10 10
加载MiXCR格式数据
MiXCR 介绍
MiXCR是一种通用的免疫组库分析软件,可以用于从任何类型的测序数据中快速准确地提取 T 细胞和 B 细胞的受体库。它处理配对和单端测序数据,考虑序列质量,纠正 PCR 错误并识别种系超突变。该软件支持部分和全长分析,并使用所有可用的 RNA 或 DNA 信息,包括 V 基因片段上游和 J 基因片段下游的序列。
准备 MiXCR 数据
MiXCR 支持以下格式的测序数据:fasta、fastq、fastq.gz、双端 fastq 和 fastq.gz。在本教程中,我使用了来自此处的真实 IGH 数据。
您可以选择使用analyze amplicon一种方式进行一次处理:
> mixcr analyze amplicon--species hs \--starting-material dna \--5-end v-primers \--3-end j-primers \--adapters adapters-present \--receptor-type IGH \input_R1.fastq input_R2.fastq analysis
或单独地执行每个步骤align,assemble和exportClones。
> mixcr align -s hs -OvParameters.geneFeatureToAlign=VTranscript \--report analysis.report input_R1.fastq input_R2.fastq analysis.vdjcaAnalysis Date: Mon Aug 25 15:22:39 MSK 2014Input file(s): input_r1.fastq,input_r2.fastqOutput file: alignments.vdjcaCommand line arguments: align --report alignmentReport.log input_r1.fastq input_r2.fastq alignments.vdjcaTotal sequencing reads: 323248Successfully aligned reads: 210360Successfully aligned, percent: 65.08%Alignment failed because of absence of V hits: 4.26%Alignment failed because of absence of J hits: 30.19%Alignment failed because of low total score: 0.48%
准备输入文件
运行完这些命令后,您将生成以下文件,其中包含有关计算出的克隆型的详细信息:
.├── analysis.clonotypes.<chains>.txt <-- This contains the count data we want!├── analysis.clna <- Build clonotypes correct PCR and sequencing errors├── analysis.vdjca <- Align raw sequences to reference sequences of segments (V, D, J) of IGH gene├── analysis.report <- Information on the run
接下来,我们将创建一个仅包含运行中指定的克隆型文件的新文件夹,并按以下格式创建一个 metadata.txt 文件。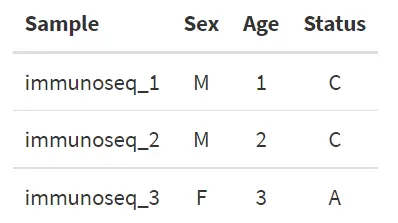
元数据文件“metadata.txt”必须是制表符分隔的文件,第一列名为“Sample”,并且有任意数量的具有任意名称的附加列。第一列应包含文件夹中没有扩展名的文件的基本名称。
加载到Immunarch包中
我们可以使用repLoad函数加载已准备好的MiXCR格式文件。
加载单个文件
# 1.1) Load the package into R:library(immunarch)# 1.2) Replace with the path to your clonotypes filefile_path = "path/to/your/mixcr/data/analysis.clonotypes.IGH.txt"# 1.3) Load MiXCR data with repLoadimmdata_mixcr <- repLoad(file_path)== Step 1/3: loading repertoire files... ==Processing "<initial>" ...-- Parsing "/path/to/your/mixcr/data/analysis.clonotypes.IGH.txt" -- mixcr== Step 2/3: checking metadata files and merging... ==Processing "<initial>" ...-- Metadata file not found; creating a dummy metadata...== Step 3/3: splitting data by barcodes and chains... ==Done!
加载成功后我们就可以查看相关文件的信息
r$> immdata_mixcr$data$data$analysis.clonotypes.IGH# A tibble: 33,812 x 15Clones Proportion CDR3.nt CDR3.aa V.name D.name J.name V.end D.start D.end J.start VJ.ins VD.ins DJ.ins Sequence<dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <int> <int> <int> <int> <int> <int> <int> <chr>1 230 0.00284 TGTGTGAGACATAAACC… CVRHKPMVQ… IGHV4-39 IGHD3-10… IGHJ6 12 NA 5 36 9 3 6 TGTGTGAGACATAAACC…2 201 0.00248 TGTGCGATTTGGGATGT… CAIWDVGLR… IGHV4-34 IGHD2-21 IGHJ4… 7 NA 5 29 10 7 3 TGTGCGATTTGGGATGT…3 179 0.00221 TGTGCGAGAGATCATGC… CARDHAGFG… IGHV1-69… IGHD3-10 IGHJ6 13 NA 4 40 18 5 13 TGTGCGAGAGATCATGC…4 99 0.00122 TGTGCGAGATGGGGATA… CARWGYCIN… IGHV4-39 IGHD2-8 IGHJ6 9 NA 6 64 23 2 21 TGTGCGAGATGGGGATA…5 97 0.00120 TGTGCGAGAGGCCCCAC… CARGPTSSE… IGHV4-34 IGHD3-22… IGHJ6 13 NA 6 52 26 24 2 TGTGCGAGAGGCCCCAC…6 97 0.00120 TGTGCGCACCACTATAC… CAHHYTSDY… IGHV2-5 IGHD1-26 IGHJ5 9 NA 2 39 19 NA 20 TGTGCGCACCACTATAC…7 92 0.00114 TGTGCGAGAGGCCCTCC… CARGPPSMG… IGHV4-34 IGHD5-24… IGHJ4 13 NA 3 38 11 6 5 TGTGCGAGAGGCCCTCC…8 84 0.00104 TGTGCGAGGTGGCTTGG… CARWLGEDI… IGHV4-39 IGHD3-16… IGHJ4… 8 NA 6 32 13 4 9 TGTGCGAGGTGGCTTGG…9 83 0.00103 TGTGCGAGAGGCCGCAG… CARGRSGDP… IGHV4-34 IGHD2-2,… IGHJ5 13 NA 4 50 18 13 5 TGTGCGAGAGGCCGCAG…10 81 0.00100 TGTGTGAGTCACCTCCT… CVSHLLDTS… IGHV1-2 IGHD2-21… IGHJ4… 8 NA 3 40 20 14 6 TGTGTGAGTCACCTCCT…# … with 33,802 more rows$meta# A tibble: 1 x 1Sample<chr>1 analysis.clonotypes.IGH
加载整个文件夹
在本教程中,我使用了三个相同的示例来显示输出,但是您应该将所有输出的.txt克隆型文件与您的metadata.txt文件一起放在此文件夹中。
# 1.1) Load the package into R:library(immunarch)# 1.2) Replace with the path to the folder with your processed MiXCR data.file_path = "/path/to/your/mixcr/data/"# 1.3) Load MiXCR data with repLoadimmdata_mixcr <- repLoad(file_path)== Step 1/3: loading repertoire files... ==Processing "/path/to/your/mixcr/data/" ...-- Parsing "/path/to/your/mixcr/data/analysis.clonotypes.IGH_1.txt" -- mixcr-- Parsing "/path/to/your/mixcr/data/analysis.clonotypes.IGH_2.txt" -- mixcr-- Parsing "/path/to/your/mixcr/data/analysis.clonotypes.IGH_3.txt" -- mixcr-- Parsing "/path/to/your/mixcr/data/metadata.txt" -- metadata== Step 2/3: checking metadata files and merging files... ==Processing "/path/to/your/mixcr/data/" ...-- Everything is OK!== Step 3/3: processing paired chain data... ==Done!
Now let’s take a look at the data! Your output should look something like below.
r$> immdata_mixcr$data$data$analysis.clonotypes.IGH_1# A tibble: 32,744 x 15Clones Proportion CDR3.nt CDR3.aa V.name D.name J.name V.end D.start D.end J.start VJ.ins VD.ins DJ.ins Sequence<dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <int> <int> <int> <int> <int> <int> <int> <chr>1 230 0.00284 TGTGTGAGACATAAACCTATGG… CVRHKPMVQG… IGHV4-39 IGHD3-10, … IGHJ6 12 NA 5 36 9 3 6 TGTGTGAGACATAAACCTATG…2 201 0.00248 TGTGCGATTTGGGATGTGGGAC… CAIWDVGLRH… IGHV4-34 IGHD2-21 IGHJ4,… 7 NA 5 29 10 7 3 TGTGCGATTTGGGATGTGGGA…3 179 0.00221 TGTGCGAGAGATCATGCGGGGT… CARDHAGFGK… IGHV1-69… IGHD3-10 IGHJ6 13 NA 4 40 18 5 13 TGTGCGAGAGATCATGCGGGG…4 99 0.00122 TGTGCGAGATGGGGATATTGTA… CARWGYCING… IGHV4-39 IGHD2-8 IGHJ6 9 NA 6 64 23 2 21 TGTGCGAGATGGGGATATTGT…5 97 0.00120 TGTGCGAGAGGCCCCACGAGCA… CARGPTSSEW… IGHV4-34 IGHD3-22, … IGHJ6 13 NA 6 52 26 24 2 TGTGCGAGAGGCCCCACGAGC…6 97 0.00120 TGTGCGCACCACTATACCAGCG… CAHHYTSDYY… IGHV2-5 IGHD1-26 IGHJ5 9 NA 2 39 19 NA 20 TGTGCGCACCACTATACCAGC…7 92 0.00114 TGTGCGAGAGGCCCTCCGTCGA… CARGPPSMGT… IGHV4-34 IGHD5-24, … IGHJ4 13 NA 3 38 11 6 5 TGTGCGAGAGGCCCTCCGTCG…8 84 0.00104 TGTGCGAGGTGGCTTGGGGAAG… CARWLGEDIR… IGHV4-39 IGHD3-16, … IGHJ4,… 8 NA 6 32 13 4 9 TGTGCGAGGTGGCTTGGGGAA…9 83 0.00103 TGTGCGAGAGGCCGCAGCGGCG… CARGRSGDPY… IGHV4-34 IGHD2-2, I… IGHJ5 13 NA 4 50 18 13 5 TGTGCGAGAGGCCGCAGCGGC…10 81 0.00100 TGTGTGAGTCACCTCCTCGACA… CVSHLLDTSD… IGHV1-2 IGHD2-21, … IGHJ4,… 8 NA 3 40 20 14 6 TGTGTGAGTCACCTCCTCGAC…# … with 32,734 more rows$data$analysis.clonotypes.IGH_2# A tibble: 32,744 x 15Clones Proportion CDR3.nt CDR3.aa V.name D.name J.name V.end D.start D.end J.start VJ.ins VD.ins DJ.ins Sequence<dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <int> <int> <int> <int> <int> <int> <int> <chr>1 230 0.00284 TGTGTGAGACATAAACCTATGG… CVRHKPMVQG… IGHV4-39 IGHD3-10, … IGHJ6 12 NA 5 36 9 3 6 TGTGTGAGACATAAACCTATG…2 201 0.00248 TGTGCGATTTGGGATGTGGGAC… CAIWDVGLRH… IGHV4-34 IGHD2-21 IGHJ4,… 7 NA 5 29 10 7 3 TGTGCGATTTGGGATGTGGGA…3 179 0.00221 TGTGCGAGAGATCATGCGGGGT… CARDHAGFGK… IGHV1-69… IGHD3-10 IGHJ6 13 NA 4 40 18 5 13 TGTGCGAGAGATCATGCGGGG…4 99 0.00122 TGTGCGAGATGGGGATATTGTA… CARWGYCING… IGHV4-39 IGHD2-8 IGHJ6 9 NA 6 64 23 2 21 TGTGCGAGATGGGGATATTGT…5 97 0.00120 TGTGCGAGAGGCCCCACGAGCA… CARGPTSSEW… IGHV4-34 IGHD3-22, … IGHJ6 13 NA 6 52 26 24 2 TGTGCGAGAGGCCCCACGAGC…6 97 0.00120 TGTGCGCACCACTATACCAGCG… CAHHYTSDYY… IGHV2-5 IGHD1-26 IGHJ5 9 NA 2 39 19 NA 20 TGTGCGCACCACTATACCAGC…7 92 0.00114 TGTGCGAGAGGCCCTCCGTCGA… CARGPPSMGT… IGHV4-34 IGHD5-24, … IGHJ4 13 NA 3 38 11 6 5 TGTGCGAGAGGCCCTCCGTCG…8 84 0.00104 TGTGCGAGGTGGCTTGGGGAAG… CARWLGEDIR… IGHV4-39 IGHD3-16, … IGHJ4,… 8 NA 6 32 13 4 9 TGTGCGAGGTGGCTTGGGGAA…9 83 0.00103 TGTGCGAGAGGCCGCAGCGGCG… CARGRSGDPY… IGHV4-34 IGHD2-2, I… IGHJ5 13 NA 4 50 18 13 5 TGTGCGAGAGGCCGCAGCGGC…10 81 0.00100 TGTGTGAGTCACCTCCTCGACA… CVSHLLDTSD… IGHV1-2 IGHD2-21, … IGHJ4,… 8 NA 3 40 20 14 6 TGTGTGAGTCACCTCCTCGAC…# … with 32,734 more rows$data$analysis.clonotypes.IGH_3# A tibble: 32,744 x 15Clones Proportion CDR3.nt CDR3.aa V.name D.name J.name V.end D.start D.end J.start VJ.ins VD.ins DJ.ins Sequence<dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <int> <int> <int> <int> <int> <int> <int> <chr>1 230 0.00284 TGTGTGAGACATAAACCTATGG… CVRHKPMVQG… IGHV4-39 IGHD3-10, … IGHJ6 12 NA 5 36 9 3 6 TGTGTGAGACATAAACCTATG…2 201 0.00248 TGTGCGATTTGGGATGTGGGAC… CAIWDVGLRH… IGHV4-34 IGHD2-21 IGHJ4,… 7 NA 5 29 10 7 3 TGTGCGATTTGGGATGTGGGA…3 179 0.00221 TGTGCGAGAGATCATGCGGGGT… CARDHAGFGK… IGHV1-69… IGHD3-10 IGHJ6 13 NA 4 40 18 5 13 TGTGCGAGAGATCATGCGGGG…4 99 0.00122 TGTGCGAGATGGGGATATTGTA… CARWGYCING… IGHV4-39 IGHD2-8 IGHJ6 9 NA 6 64 23 2 21 TGTGCGAGATGGGGATATTGT…5 97 0.00120 TGTGCGAGAGGCCCCACGAGCA… CARGPTSSEW… IGHV4-34 IGHD3-22, … IGHJ6 13 NA 6 52 26 24 2 TGTGCGAGAGGCCCCACGAGC…6 97 0.00120 TGTGCGCACCACTATACCAGCG… CAHHYTSDYY… IGHV2-5 IGHD1-26 IGHJ5 9 NA 2 39 19 NA 20 TGTGCGCACCACTATACCAGC…7 92 0.00114 TGTGCGAGAGGCCCTCCGTCGA… CARGPPSMGT… IGHV4-34 IGHD5-24, … IGHJ4 13 NA 3 38 11 6 5 TGTGCGAGAGGCCCTCCGTCG…8 84 0.00104 TGTGCGAGGTGGCTTGGGGAAG… CARWLGEDIR… IGHV4-39 IGHD3-16, … IGHJ4,… 8 NA 6 32 13 4 9 TGTGCGAGGTGGCTTGGGGAA…9 83 0.00103 TGTGCGAGAGGCCGCAGCGGCG… CARGRSGDPY… IGHV4-34 IGHD2-2, I… IGHJ5 13 NA 4 50 18 13 5 TGTGCGAGAGGCCGCAGCGGC…10 81 0.00100 TGTGTGAGTCACCTCCTCGACA… CVSHLLDTSD… IGHV1-2 IGHD2-21, … IGHJ4,… 8 NA 3 40 20 14 6 TGTGTGAGTCACCTCCTCGAC…# … with 32,734 more rows$meta# A tibble: 3 x 4Sample Sex Age Status<chr> <chr> <dbl> <chr>1 analysis.clonotypes.IGH_1 M 1 C2 analysis.clonotypes.IGH_2 M 2 C3 analysis.clonotypes.IGH_3 F 3 A
加载10x Genomics格式数据
10x Genomics简介
10x Genomics Chromium 单细胞免疫组库分析方案可同时分析以下内容:
- T 和 B 细胞的 V(D)J 转录本和克隆型。
- 5’ 基因表达。
- 同一组细胞在单细胞分辨率下的细胞表面蛋白/抗原特异性(特征条形码)。
他们的Cell Ranger综合分析软件,包括以下用于免疫组库分析的工具:
cellranger mkfastq将 Illumina 测序仪生成的原始碱基检出 (BCL) 文件解复用为 FASTQ 文件。它是 Illumina 的 bcl2fastq 的包装器,具有特定于 10x 库的附加有用功能和简化的样本表格式。cellranger vdj从 cellranger mkfastq 中获取用于 V(D)J 库的 FASTQ 文件,并执行序列组装和配对克隆型调用。它使用 Chromium 细胞条形码和 UMI 来组装每个细胞的 V(D)J 转录本。克隆型和 CDR3 序列作为 .vloupe 文件输出,可以加载到 Loupe V(D)J 浏览器中进行可视化探索。cellranger count为 5’ 基因表达和/或特征条码(细胞表面蛋白或抗原)库获取 FASTQ 文件,并执行比对、过滤、条码计数和 UMI 计数。它使用 Chromium 细胞条形码生成特征条形码矩阵、确定聚类并执行基因表达分析。cellranger 计数管道输出一个 .cloupe 文件,该文件可以加载到Loupe Browser中以进行交互式可视化、聚类和差异表达分析。
准备10x Genomics格式数据
使用cellranger vdj处理数据后,将输出很多结果文件。我们将使用filtered contigs.csv文件,该文件中包含了barcode信息。
.├── vdj_v1_mm_c57bl6_pbmc_t_filtered_contig_annotations.csv <-- 这里包含了我们想要的计数数据!├── vdj_v1_mm_c57bl6_pbmc_t_consensus_annotations.csv├── vdj_v1_mm_c57bl6_pbmc_t_clonotypes.csv├── vdj_v1_mm_c57bl6_pbmc_t_all_contig_annotations.csv├── vdj_v1_mm_c57bl6_pbmc_t_matrix.h5├── vdj_v1_mm_c57bl6_pbmc_t_bam.bam.bai├── vdj_v1_mm_c57bl6_pbmc_t_molecule_info.h5├── vdj_v1_mm_c57bl6_pbmc_t_raw_feature_bc_matrix.tar.gz├── vdj_v1_mm_c57bl6_pbmc_t_analysis.tar.gz
加载到Immunarch包中
使用repLoad函数加载整个文件夹
# 1.1) Load the package into R:library(immunarch)# 1.2) Replace with the path to your processed 10x data or to the clonotypes filefile_path = "~/path/to/your/cellranger/data/"# 1.3) Load 10x data with repLoadimmdata_10x <- repLoad(file_path)== Step 1/3: loading repertoire files... ==Processing "/filepath/C57BL_mice_igenrichment" ...-- Parsing "/filepath/vdj_v1_mm_c57bl6_pbmc_t_all_contig_annotations.csv" -- 10x (filt.contigs)[!] Removed 2917 clonotypes with no nucleotide and amino acid CDR3 sequence.-- Parsing "/filepath/vdj_v1_mm_c57bl6_pbmc_t_clonotypes.csv" -- unsupported format, skipping-- Parsing "/filepath/vdj_v1_mm_c57bl6_pbmc_t_consensus_annotations.csv" -- 10x (consensus)-- Parsing "/filepath/vdj_v1_mm_c57bl6_pbmc_t_filtered_contig_annotations.csv" -- 10x (filt.contigs)[!] Removed 1198 clonotypes with no nucleotide and amino acid CDR3 sequence.== Step 2/3: checking metadata files and merging... ==Processing "<initial>" ...-- Metadata file not found; creating a dummy metadata...== Step 3/3: splitting data by barcodes and chains... ==Done!
加载成功后,我们来查看免疫组库的相关信息。
> immdata_10x$data$vdj_v1_mm_c57bl6_splenocytes_t_consensus_annotations_TRA# A tibble: 710 x 17Clones Proportion CDR3.nt CDR3.aa V.name D.name J.name V.end D.start D.end J.start VJ.ins VD.ins DJ.ins chain ClonotypeID ConsensusID<dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <int> <int> <int> <int> <int> <int> <int> <chr> <chr> <chr>1 55 0.00414 TGTGCTATGGC… CAMATGG… TRAV13… None TRAJ56 NA NA NA NA NA NA NA TRA clonotype306 clonotype30…2 55 0.00414 TGTGCAGCTAG… CAASGNT… TRAV7-4 None TRAJ27 NA NA NA NA NA NA NA TRA clonotype338 clonotype33…3 53 0.00399 TGTGCAGCAAG… CAARDSG… TRAV14… None TRAJ11 NA NA NA NA NA NA NA TRA clonotype617 clonotype61…4 45 0.00339 TGCGCAGTCAG… CAVSNNT… TRAV3-3 None TRAJ27 NA NA NA NA NA NA NA TRA clonotype435 clonotype43…5 43 0.00324 TGTGCAGTCAG… CAVSNMG… TRAV7D… None TRAJ9 NA NA NA NA NA NA NA TRA clonotype401 clonotype40…6 42 0.00316 TGTGCAGCAAG… CAASPNY… TRAV14… None TRAJ21 NA NA NA NA NA NA NA TRA clonotype5 clonotype5_…7 37 0.00279 TGTGCAGTGAG… CAVSSGG… TRAV7D… None TRAJ6 NA NA NA NA NA NA NA TRA clonotype453 clonotype45…8 35 0.00264 TGTGCAGCAAG… CAASATS… TRAV14… None TRAJ22 NA NA NA NA NA NA NA TRA clonotype809 clonotype80…9 32 0.00241 TGTGCAGCAAG… CAASPNY… TRAV14… None TRAJ21 NA NA NA NA NA NA NA TRA clonotype150 clonotype15…10 32 0.00241 TGTGCTCTGGG… CALGDEA… TRAV6-… None TRAJ30 NA NA NA NA NA NA NA TRA clonotype393 clonotype39…# … with 700 more rows$metaSample Chain Source1 vdj_v1_mm_c57bl6_splenocytes_t_all_contig_annotations_Multi Multi vdj_v1_mm_c57bl6_splenocytes_t_all_contig_annotations2 vdj_v1_mm_c57bl6_splenocytes_t_all_contig_annotations_TRA TRA vdj_v1_mm_c57bl6_splenocytes_t_all_contig_annotations3 vdj_v1_mm_c57bl6_splenocytes_t_all_contig_annotations_TRB TRB vdj_v1_mm_c57bl6_splenocytes_t_all_contig_annotations5 vdj_v1_mm_c57bl6_splenocytes_t_consensus_annotations_TRA TRA vdj_v1_mm_c57bl6_splenocytes_t_consensus_annotations6 vdj_v1_mm_c57bl6_splenocytes_t_consensus_annotations_TRB TRB vdj_v1_mm_c57bl6_splenocytes_t_consensus_annotations
注意事项:
一个重要的注意事项是某些 contigs 文件可能缺少barcode的列 - 细胞的唯一标识。这些文件可用于分析单链数据(仅 alpha 或 beta TCR),但为了分析配对链数据并充分利用单细胞技术的全部功能,您应该将带有条形码的文件读入到Immunarch中。
参考来源:https://immunarch.com/articles/web_only/load_10x.html

若有收获,就点个赞吧
0 人点赞
