克隆型追踪 Tracking of clonotypes
克隆型追踪(Clonotype tracking)是一种用于监测疫苗接种和癌症免疫学中感兴趣的克隆型频率变化的常用方法。例如,研究人员可以追踪疫苗在接种前和接种后不同时间点的克隆型变化,或分析肿瘤样本中恶性细胞克隆型的生长情况。
在immunarch包中,我们可以使用trackClonotypes函数进行克隆型追踪分析,且分析结果可直接导入到vis函数中进行可视化展示。immunarch中集成了多种克隆型追踪方法,目前主要有三种方法可供选择。
1. 追踪最丰富的克隆型
最简单的方法,是从一个输入的免疫组库中选择最丰富的克隆类型,并批量跟踪所有的免疫组库序列。其中,参数.which和.col可用于选择免疫组库、从中获取的克隆型数量以及要使用的列。
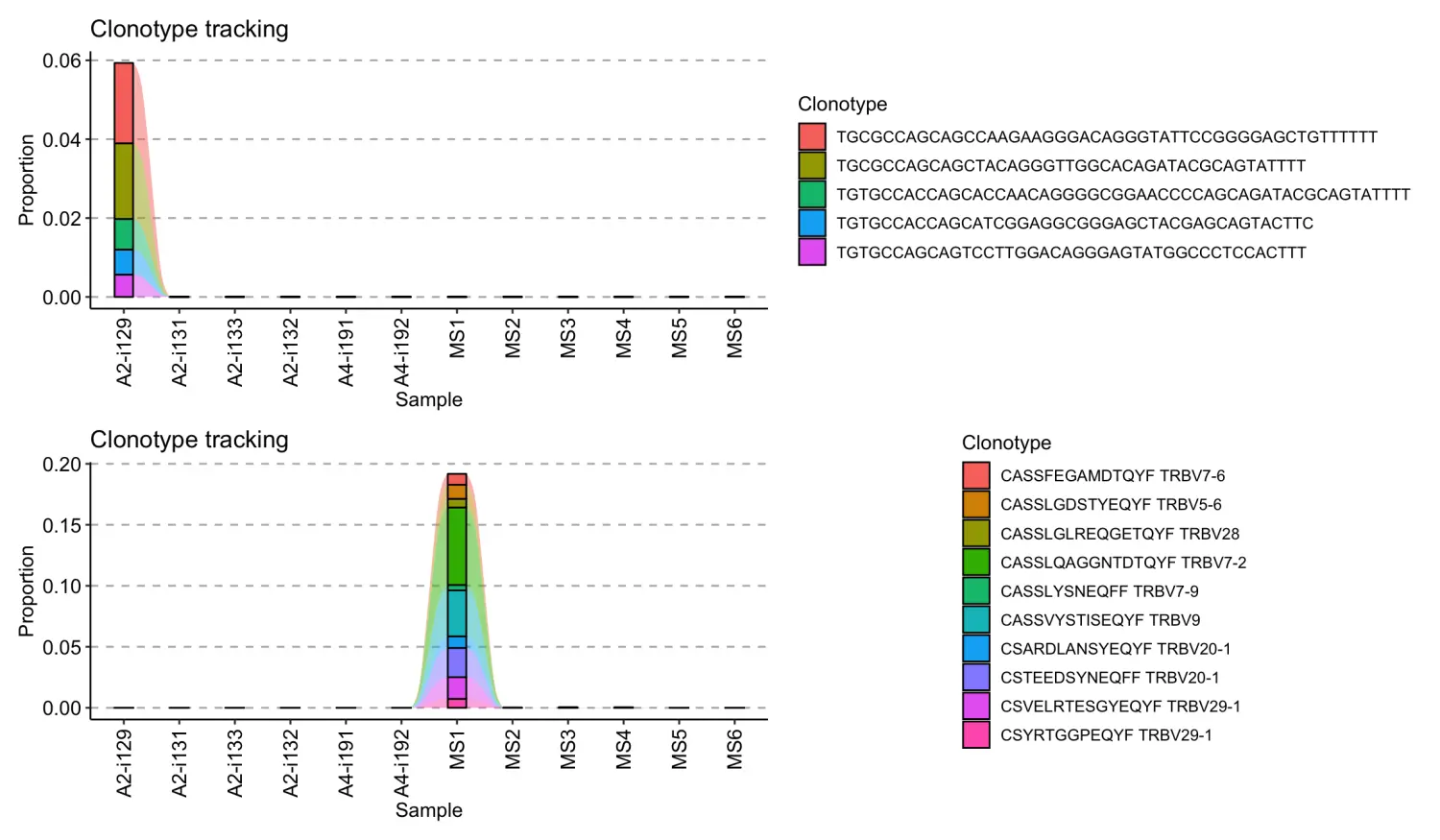
例如,我们可以从第一个库中选择10个最丰富的克隆型,并使用他们的CDR3核苷酸序列对其进行跟踪:
tc1 <- trackClonotypes(immdata$data, list(1, 5), .col = "nt")head(tc1)# CDR3.nt A2-i129 A2-i131 A2-i133 A2-i132#1: TGCGCCAGCAGCCAAGAAGGGACAGGGTATTCCGGGGAGCTGTTTTTT 0.020352941 0 0 #0#2: TGCGCCAGCAGCTACAGGGTTGGCACAGATACGCAGTATTTT 0.019176471 0 0 0#3: TGTGCCACCAGCACCAACAGGGGCGGAACCCCAGCAGATACGCAGTATTTT 0.007764706 0 #0 0#4: TGTGCCACCAGCATCGGAGGCGGGAGCTACGAGCAGTACTTC 0.006352941 0 0 0#5: TGTGCCAGCAGTCCTTGGACAGGGAGTATGGCCCTCCACTTT 0.005647059 0 0 0# A4-i191 A4-i192 MS1 MS2 MS3 MS4 MS5 MS6#1: 0 0 0 0 0 0 0 0#2: 0 0 0 0 0 0 0 0#3: 0 0 0 0 0 0 0 0#4: 0 0 0 0 0 0 0 0#5: 0 0 0 0 0 0 0 0
参数的值list(1,5)表示从immdata$data的第一个repertoire列表中选择5个clonotypes。.col = “nt”表示应只接受CDR3核苷酸序列。
从“MS1”库中选择10个最丰富的氨基酸克隆型序列及其V基因进行跟踪:
tc2 <- trackClonotypes ( immdata $ data , list ( "MS1" , 10 ), .col = "aa+v" )head(tc2)# CDR3.aa V.name A2-i129 A2-i131 A2-i133 A2-i132 A4-i191 A4-i192 MS1#1: CASSFEGAMDTQYF TRBV7-6 0 0 0 0 0 0 0.008941176#2: CASSLGDSTYEQYF TRBV5-6 0 0 0 0 0 0 0.011529412#3: CASSLGLREQGETQYF TRBV28 0 0 0 0 0 0 0.007058824#4: CASSLQAGGNTDTQYF TRBV7-2 0 0 0 0 0 0 0.063529412#5: CASSLYSNEQFF TRBV7-9 0 0 0 0 0 0 0.004470588#6: CASSVYSTISEQYF TRBV9 0 0 0 0 0 0 0.037647059# MS2 MS3 MS4 MS5 MS6#1: 0.0000000000 0.0000000000 0.0000000000 0 0#2: 0.0000000000 0.0000000000 0.0001176471 0 0#3: 0.0001176471 0.0000000000 0.0000000000 0 0#4: 0.0000000000 0.0000000000 0.0000000000 0 0#5: 0.0000000000 0.0000000000 0.0000000000 0 0#6: 0.0000000000 0.0001176471 0.0001176471 0 0
参数的值list("MS1",10)表示从immdata$data的”MS1”免疫组库中选择10个克隆型,.col=“aa+v”表示应该同时选取CDR3氨基酸序列和最丰富的克隆型的v基因片段。
我们可以使用两种不同的方法进行结果的可视化:
p1 <- vis ( tc1 )p2 <- vis ( tc2 )p1 / p2
2. 追踪具有特定核苷酸或氨基酸序列的克隆型
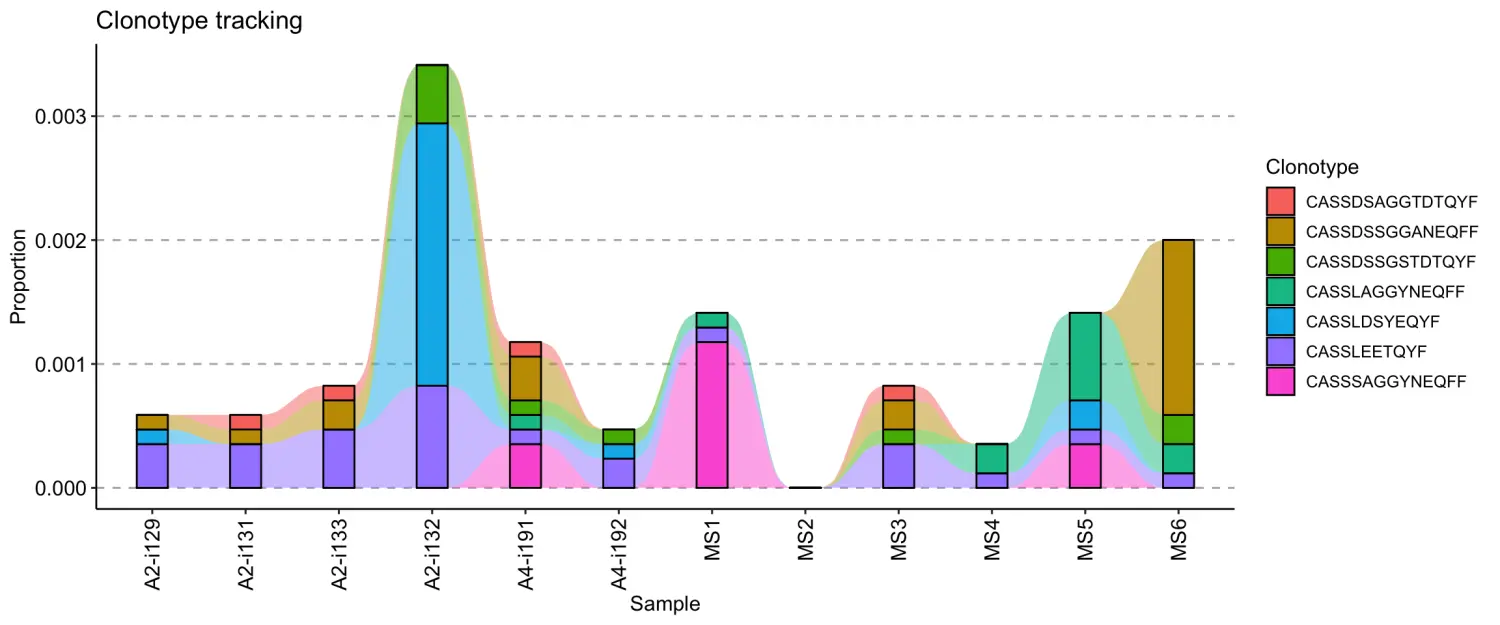
为了追踪特定的克隆型序列,我们可以通过.which参数指定特定的核苷酸或氨基酸序列,同时提供.col参数指定在哪些列中搜索序列。例如,要追踪下面指定的七个 CDR3 氨基酸序列,您需要执行以下代码:
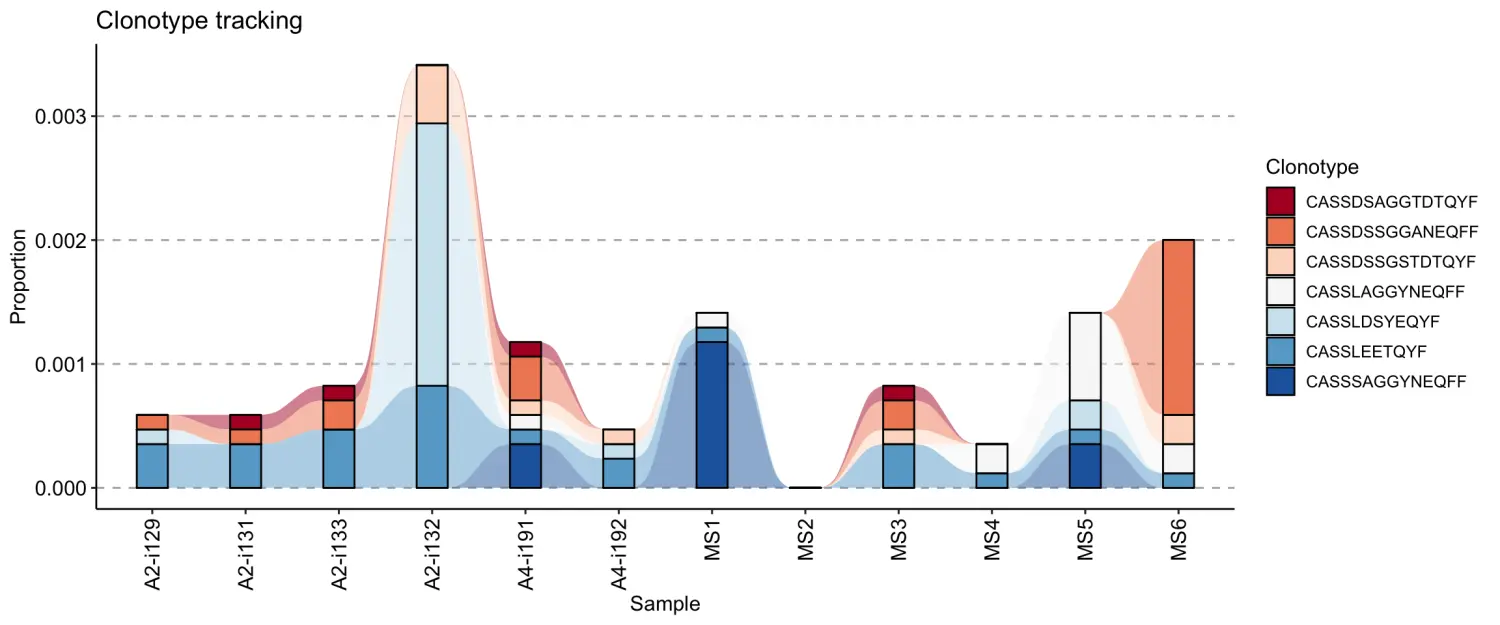
target <- c("CASSLEETQYF", "CASSDSSGGANEQFF", "CASSDSSGSTDTQYF", "CASSLAGGYNEQFF", "CASSDSAGGTDTQYF", "CASSLDSYEQYF", "CASSSAGGYNEQFF")tc <- trackClonotypes(immdata$data, target, .col = "aa")vis(tc)
3. 追具有特定序列和基因片段的克隆型
同样的,我们还可以使用有关序列和基因片段的信息来追踪克隆型的变化。输入一个具有特定 CDR3 序列和基因片段的序列数据框。我们将通过从所有免疫组库的第一个库中选择 10 个最丰富的克隆型来模拟这一点:
target <- immdata$data[[1]] %>%select(CDR3.aa, V.name) %>%head(10)target## # A tibble: 10 x 2## CDR3.aa V.name## <chr> <chr>## 1 CASSQEGTGYSGELFF TRBV4-1## 2 CASSYRVGTDTQYF TRBV4-1## 3 CATSTNRGGTPADTQYF TRBV15## 4 CATSIGGGSYEQYF TRBV15## 5 CASSPWTGSMALHF TRBV27## 6 CASQGDSFNSPLHF TRBV4-1## 7 CASSQDMGGRNTGELFF TRBV4-1## 8 CASSEEPRLFGYTF TRBV2## 9 CASSQPGQGGGDEQFF TRBV4-1## 10 CASSWVARGPYEQYF TRBV6-6
接下来,我们将此数据框作为参数提供给.which参数以追踪目标克隆型的变化:
tc <- trackClonotypes(immdata$data, target)vis(tc)
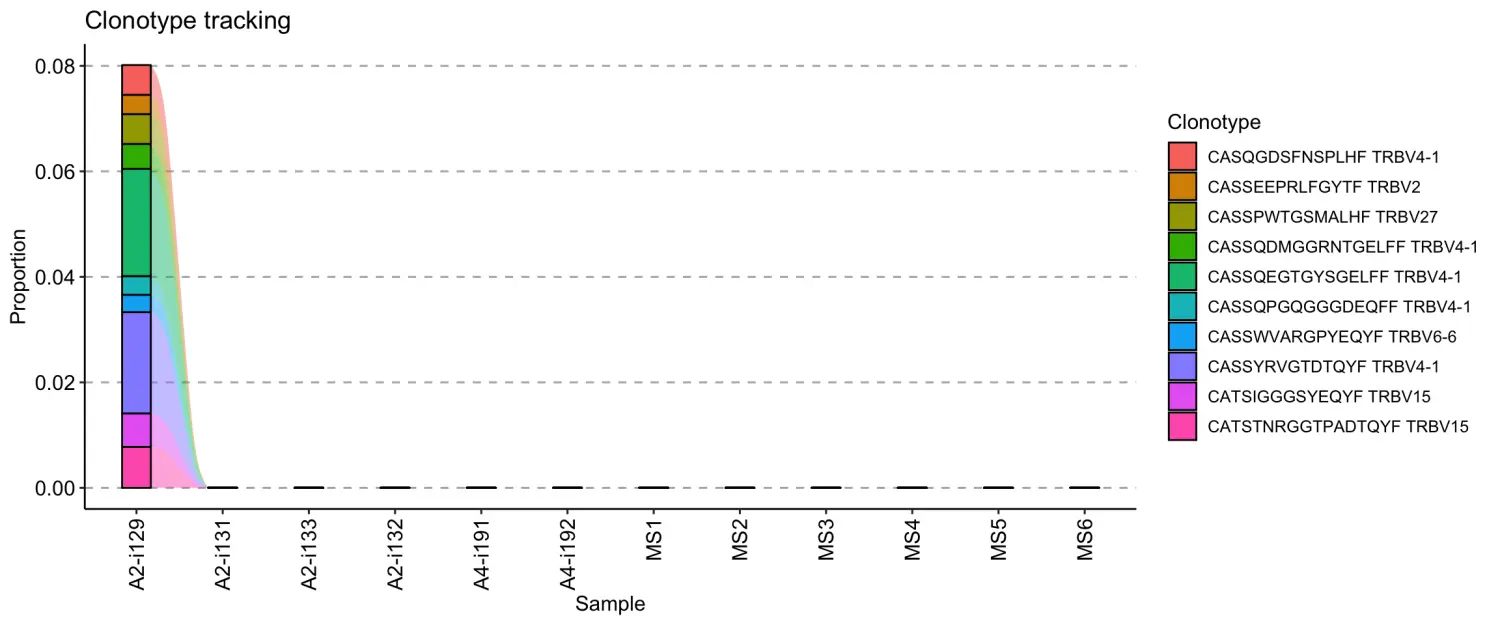
请注意,我们可以使用target数据框中的任何列,例如 CDR3 核苷酸和氨基酸序列以及任何基因片段。
克隆型追踪可视化
在immunarch包中,共提供了三种可视化克隆型跟踪的方法。要选择绘图的类型,您需要为函数提供".plot"参数`指定三种绘图类型之一:
.plot = "smooth"- 默认使用,使用平滑线和堆积条形图的可视化;
-.plot = "area"- 使用丰度线下的区域可视化丰度;
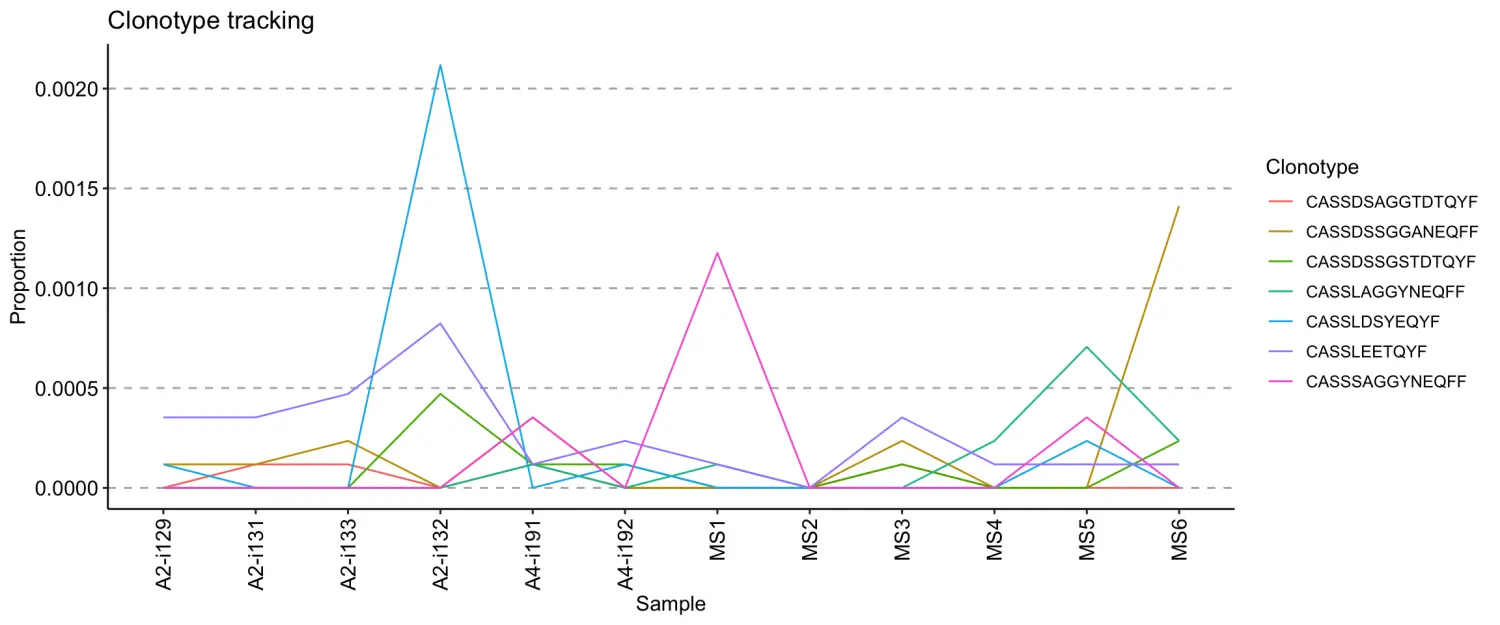
-.plot = "line"- 仅可视化线条,连接不同时间点之间同一克隆型的丰度水平。
target <- c("CASSLEETQYF", "CASSDSSGGANEQFF", "CASSDSSGSTDTQYF", "CASSLAGGYNEQFF", "CASSDSAGGTDTQYF", "CASSLDSYEQYF", "CASSSAGGYNEQFF")tc <- trackClonotypes(immdata$data, target, .col = "aa")vis(tc, .plot = "smooth")
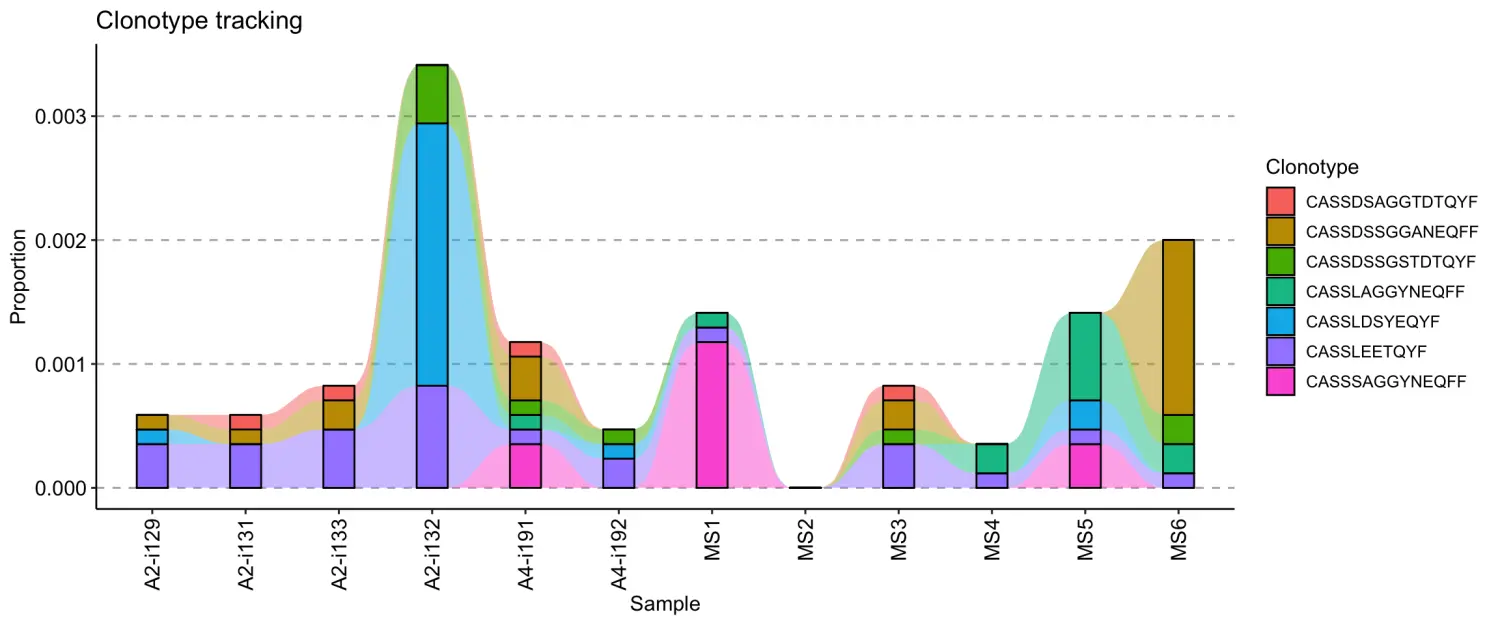
vis(tc, .plot = "area")
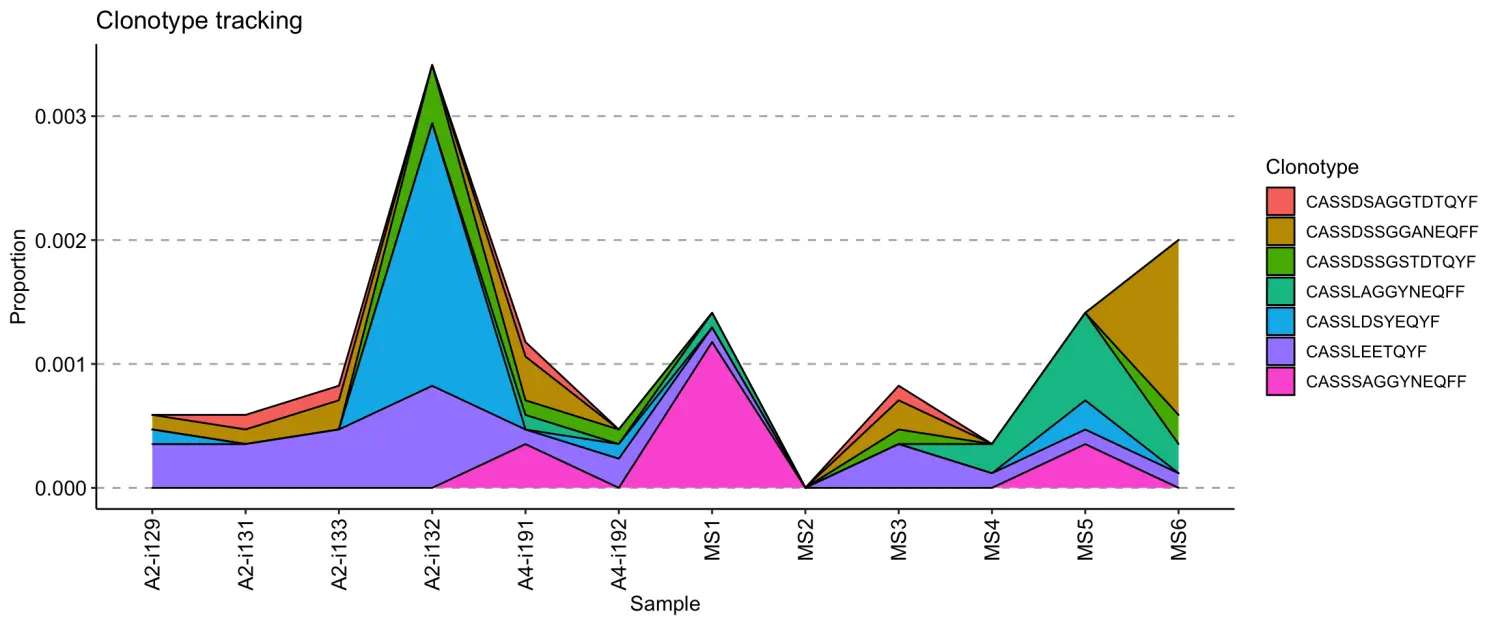
vis(tc, .plot = "line")
更改样本顺序
我们可以通过.order参数控制可视化图形中的样本顺序。您可以传递您计划可视化的样本索引或样本名称。
# Passing indicesnames(immdata$data)[c(1, 3, 5)] # check sample names## [1] "A2-i129" "A2-i133" "A4-i191"vis(tc, .order = c(1, 3, 5))
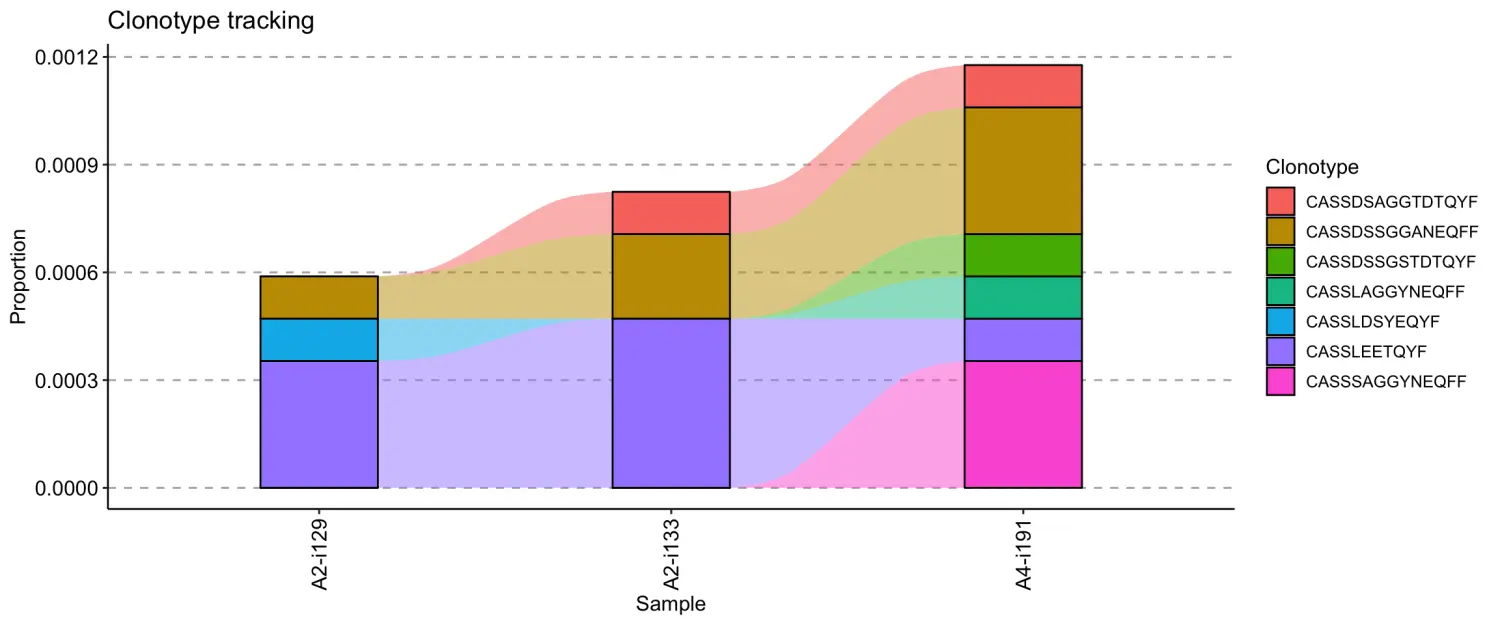
# You can change the ordervis(tc, .order = c(5, 1, 3))
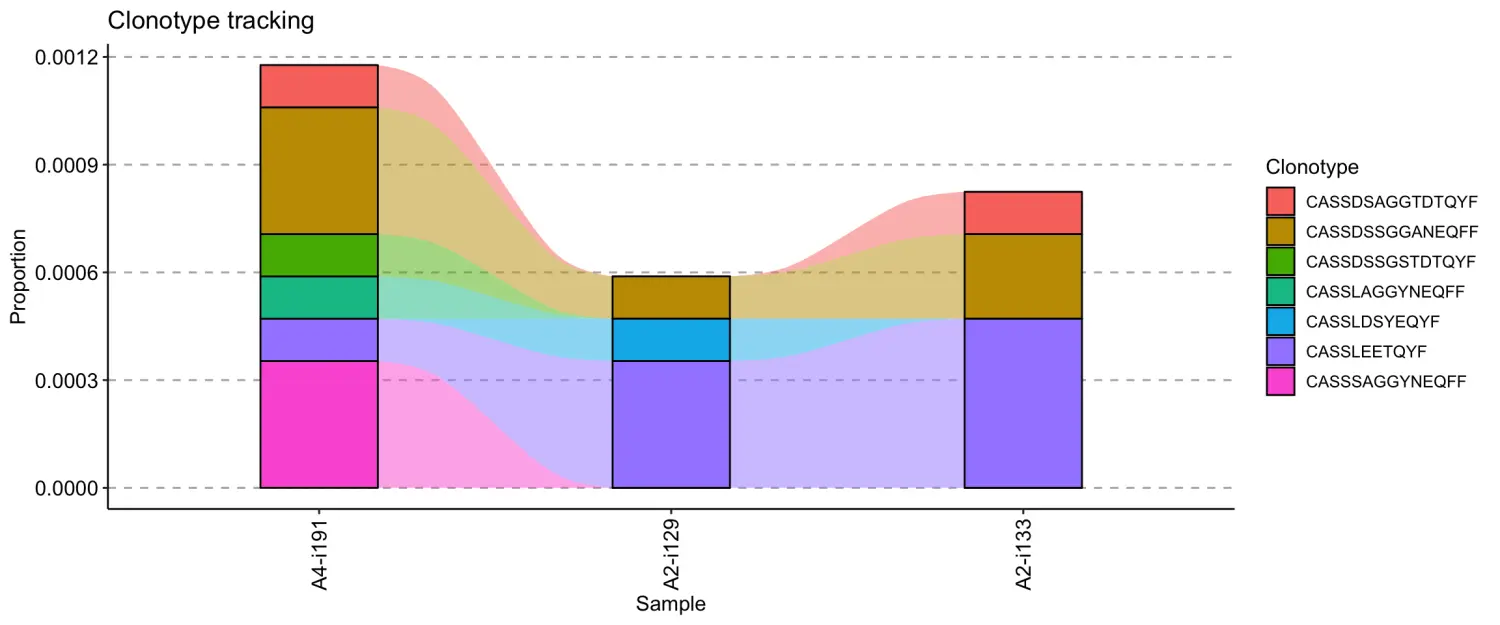
# Passing sample namesvis(tc, .order = c("A2-i129", "A2-i133", "A4-i191"))
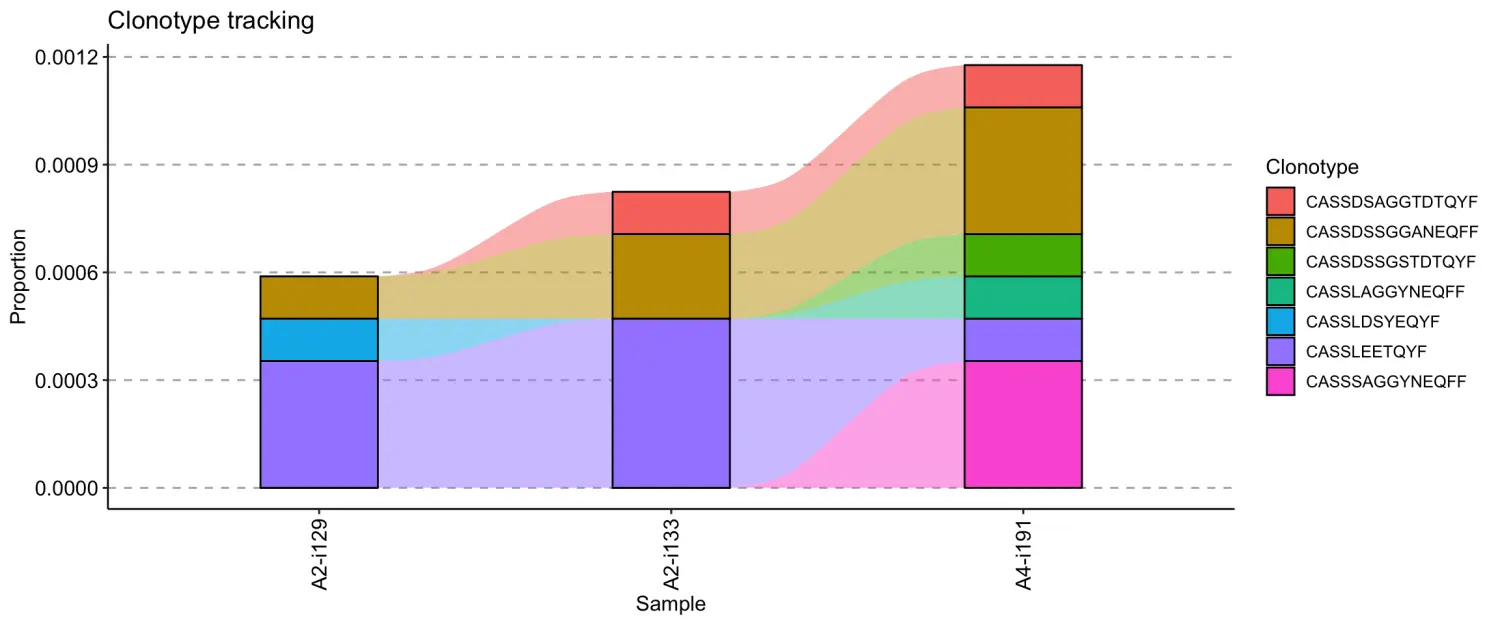
如果我们的元数据(metadata)中包含了有关时间的信息,如接种疫苗或肿瘤样本的时间点,则可以使用它相应地对样本重新排序。在我们的示例中,immdata$meta不包含关于时间点的信息,因此我们将模拟这种情况。
immdata$meta$Timepoint <- sample(1:length(immdata$data))immdata$meta## # A tibble: 12 x 7## Sample ID Sex Age Status Lane Timepoint## <chr> <chr> <chr> <dbl> <chr> <chr> <int>## 1 A2-i129 C1 M 11 C A 12## 2 A2-i131 C2 M 9 C A 11## 3 A2-i133 C4 M 16 C A 8## 4 A2-i132 C3 F 6 C A 2## 5 A4-i191 C8 F 22 C B 1## 6 A4-i192 C9 F 24 C B 10## 7 MS1 MS1 M 12 MS C 7## 8 MS2 MS2 M 30 MS C 6## 9 MS3 MS3 M 8 MS C 5## 10 MS4 MS4 F 14 MS C 9## 11 MS5 MS5 F 15 MS C 3## 12 MS6 MS6 F 15 MS C 4
接下来,我们根据“Timepoint”列(从最小到最大),以正确的顺序创建一个包含样本的向量:
sample_order <- order(immdata$meta$Timepoint)
排序后,时间点遵循正确的顺序:
immdata$meta$Timepoint[sample_order]## [1] 1 2 3 4 5 6 7 8 9 10 11 12
样本按时间点排序:
immdata$meta$Sample[sample_order]## [1] "A4-i191" "A2-i132" "MS5" "MS6" "MS3" "MS2" "MS1"## [8] "A2-i133" "MS4" "A4-i192" "A2-i131" "A2-i129"
最后,我们将数据进行可视化:
vis(tc, .order = sample_order)
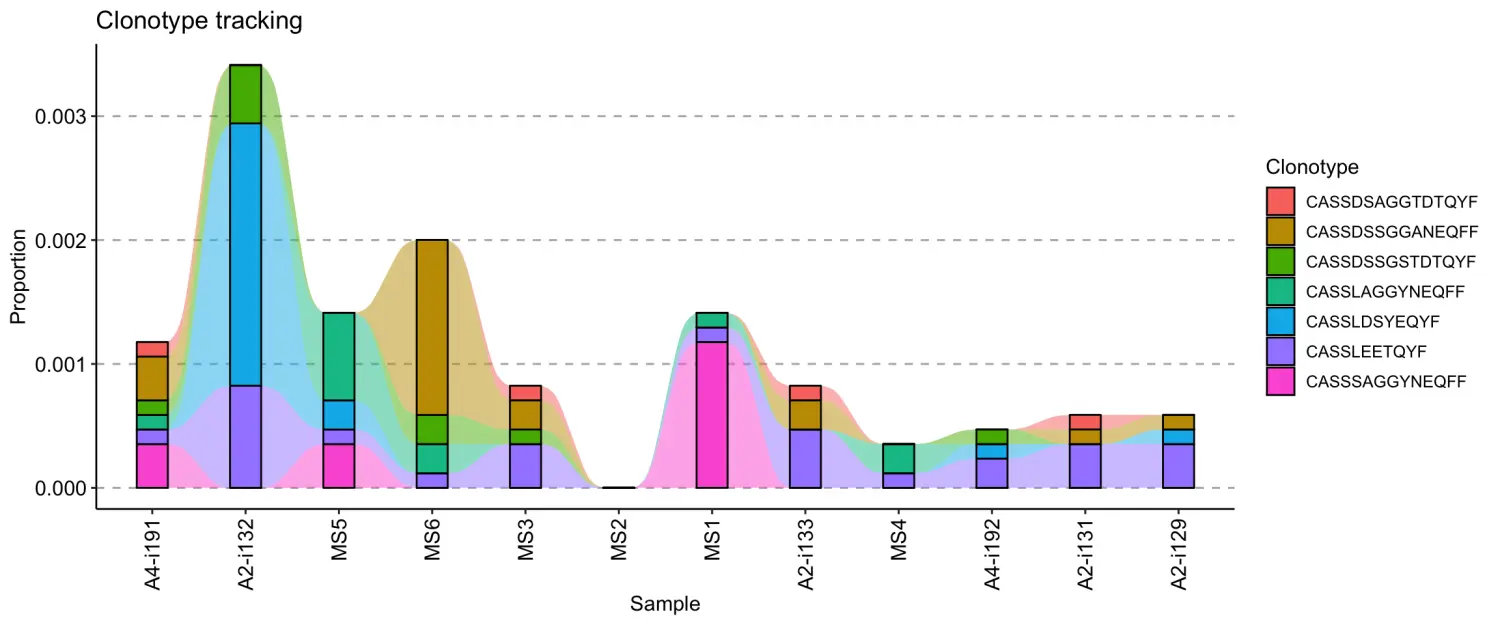
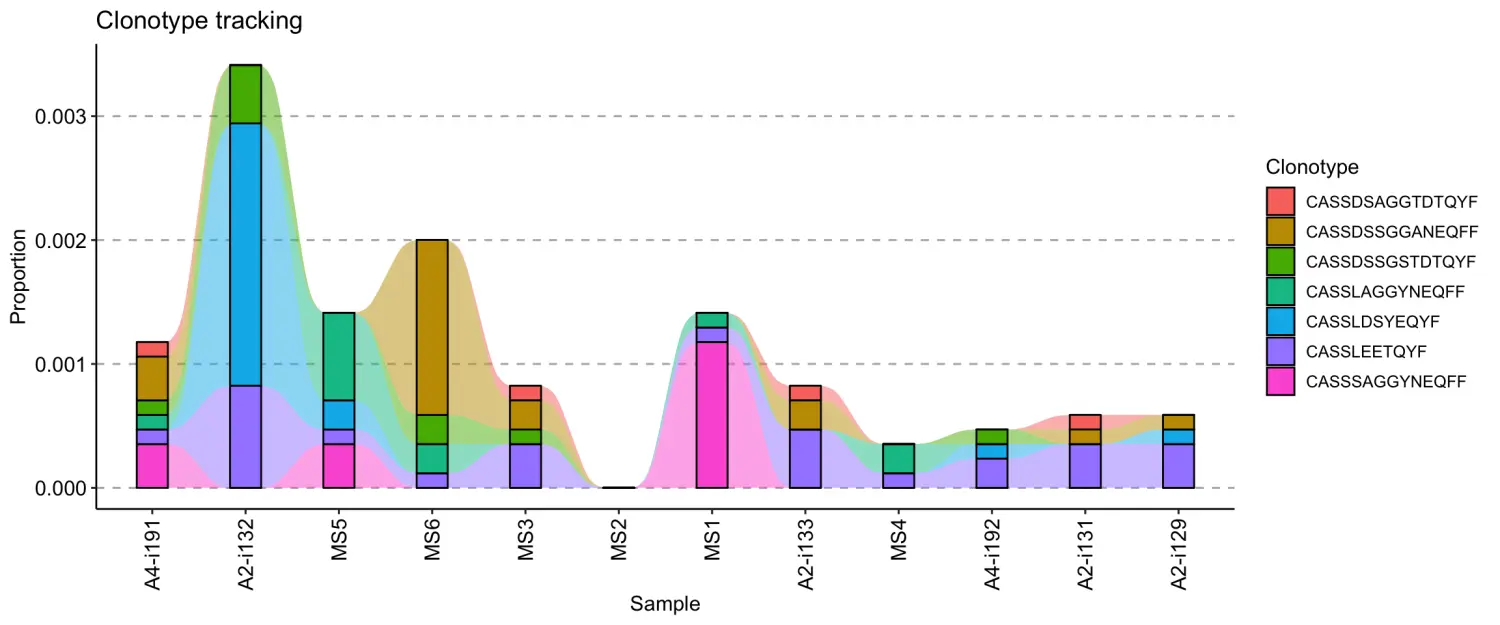
可以使用单行命令完成以上操作:
vis(tc, .order = order(immdata$meta$Timepoint))
更改调色板
如果要更改调色板,我们可以添加 ggplot2函数的scalefill*参数,在 R 控制台中运行?scale_fill_brewer以了解有关 ColorBrewer 及其配色方案的更多信息。建议使用scale_fill_brewer:
vis(tc) + scale_fill_brewer(palette = "Spectral")
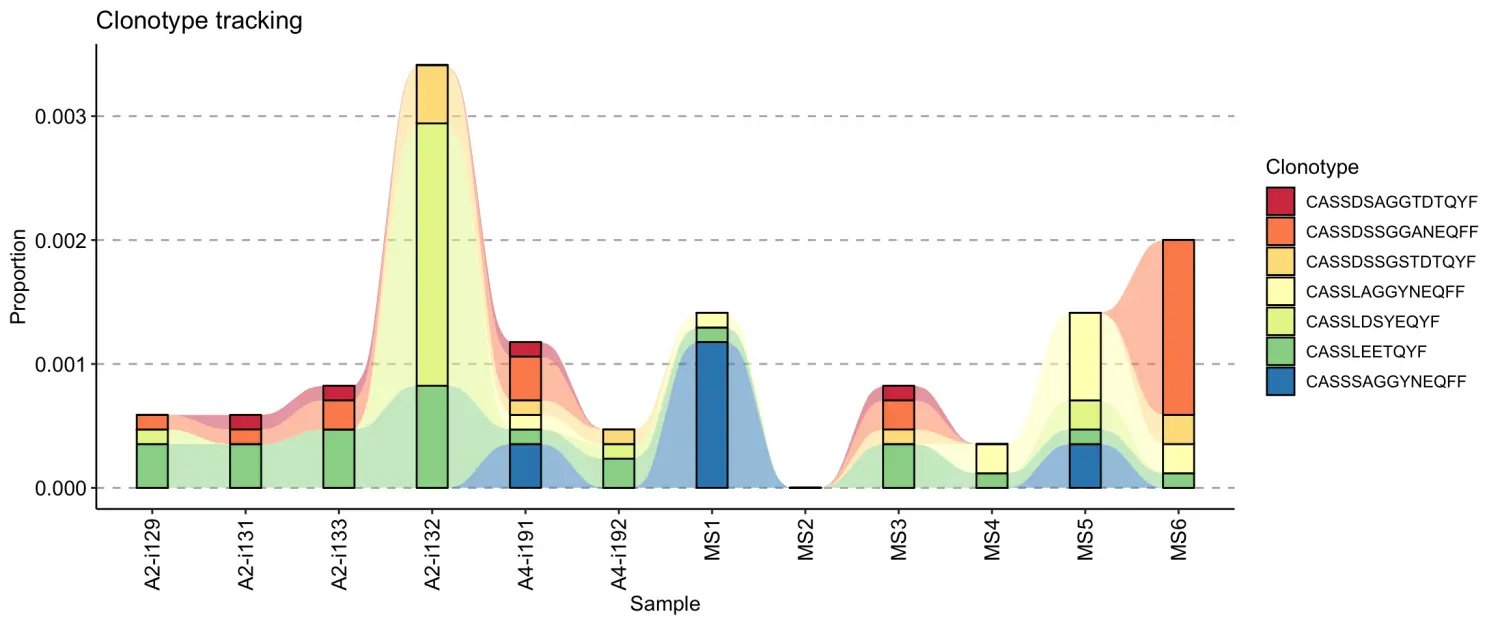
vis(tc) + scale_fill_brewer(palette = "RdBu")
参考来源:https://immunarch.com/articles/web_only/v8_tracking.html

若有收获,就点个赞吧
0 人点赞
