基因使用计算
immunarch包带有一个基因片段数据表,其中包含了遵循IMGT命名法的几个物种的已知VDJ基因片段。我们可以调用gene_stats()函数查看基因的当前统计信息::
gene_stats()## alias species ighd ighj ighv igij igkj igkv iglj iglv traj## 1 bt BosTaurus 21 4 25 0 1 6 5 26 46## 2 cd CamelusDromedarius 0 0 0 0 0 0 0 0 0## 3 clf CanisLupusFamiliaris 0 0 0 0 0 0 0 0 0## 4 dr DanioRerio 7 7 0 3 0 0 0 0 0## 5 hs HomoSapiens 30 13 248 0 5 64 7 69 57## 6 macmul MacacaMulatta 24 7 19 0 4 83 5 0 0## 7 mmc MusMusculusCastaneus 0 0 0 0 0 4 0 0 0## 8 mmd MusMusculusDomesticus 0 0 0 0 0 2 0 0 0## 9 musmus MusMusculus 32 8 225 0 8 109 3 5 42## 10 oa OrnithorhynchusAnatinus 3 10 0 0 0 0 0 0 0## 11 oc OryctolagusCuniculus 10 11 39 0 8 26 2 20 0## 12 om OncorhynchusMykiss 9 7 6 0 0 0 0 0 0## 13 rn RattusNorvegicus 30 4 113 0 6 132 2 8 0## 14 smth MusMusculusMolossinus 0 0 0 0 0 1 0 0 0## 15 smth MusMusculusMusculus 0 0 0 0 0 1 0 0 0## 16 smth MusSpretus 0 0 0 0 0 2 0 2 0## 17 ss SusScrofa 5 5 15 0 8 19 4 14 0## trav trbd trbj trbv trdd trdj trdv trgj trgv## 1 0 0 0 0 5 3 0 6 15## 2 0 0 0 0 0 0 7 2 2## 3 0 2 8 19 0 0 0 7 8## 4 0 0 0 0 0 0 0 0 0## 5 60 3 14 64 3 4 6 4 10## 6 0 2 15 58 0 0 0 0 0## 7 0 0 0 0 0 0 0 0 0## 8 0 0 0 0 0 0 0 0 0## 9 145 2 14 23 2 3 7 0 11## 10 0 0 0 0 0 0 0 0 0## 11 0 0 0 0 0 0 0 0 0## 12 0 1 9 0 0 0 0 0 0## 13 0 0 0 0 0 0 0 0 0## 14 0 0 0 0 0 0 0 0 0## 15 0 0 0 0 0 0 0 0 0## 16 0 0 0 0 0 0 0 0 0## 17 0 0 0 0 0 0 0 0 0
为了计算不同VDJ基因的分布情况,immunarch提供了geneUsage函数。它接收一个或多个免疫组库列表作为输入,以及您想要获取统计数据的基因和物种。例如,如果您打算计算Homo Sapiens的TRBV基因,则需要使用hs.trbv参数,其中hs是物种别名,trbv是基因名称。如果您想要就算Mus Musculus的IGHJ基因,则需要使用musmus.ighj参数:
# Next four function calls are equal. "hs" is from the "alias" column.imm_gu <- geneUsage(immdata$data, "hs.trbv")# imm_gu = geneUsage(immdata$data, "HomoSapiens.trbv")# imm_gu = geneUsage(immdata$data, "hs.TRBV")# imm_gu = geneUsage(immdata$data, "HomoSapiens.TRBV")imm_gu## # A tibble: 48 x 13## Names `A2-i129` `A2-i131` `A2-i133` `A2-i132` `A4-i191` `A4-i192` MS1 MS2## <chr> <int> <int> <int> <int> <int> <int> <int> <int>## 1 TRBV… 24 28 NA 16 29 6 19 4## 2 TRBV… 42 60 8 29 28 16 25 35## 3 TRBV… 230 282 135 108 376 215 195 142## 4 TRBV… 21 14 26 17 17 16 14 10## 5 TRBV… 183 172 125 161 95 113 94 105## 6 TRBV… 8 11 5 24 2 7 4 13## 7 TRBV… 603 459 313 433 333 557 406 606## 8 TRBV… 37 54 8 38 18 17 7 17## 9 TRBV… 44 53 45 78 29 43 39 28## 10 TRBV… 65 91 48 73 40 30 23 94## # … with 38 more rows, and 4 more variables: MS3 <int>, MS4 <int>, MS5 <int>,## # MS6 <int>
我们还可以通过.quant参数使用单个克隆型的计数 ( .quant = "count") 或不使用它们 ( .quant = NA)来计算基因的分布。
为了计算等位基因级别(allele-level )或家族级别(family-level)的分布,我们还可以更改.type参数。
设置.norm参数控制是否将数据进行归一化,以确保所有频率之和等于 1。
您可以通过不同方式可视化基因使用频率的直方图:
# Compute the distribution of the first two samplesimm_gu <- geneUsage(immdata$data[c(1, 2)], "hs.trbv", .norm = T)vis(imm_gu)
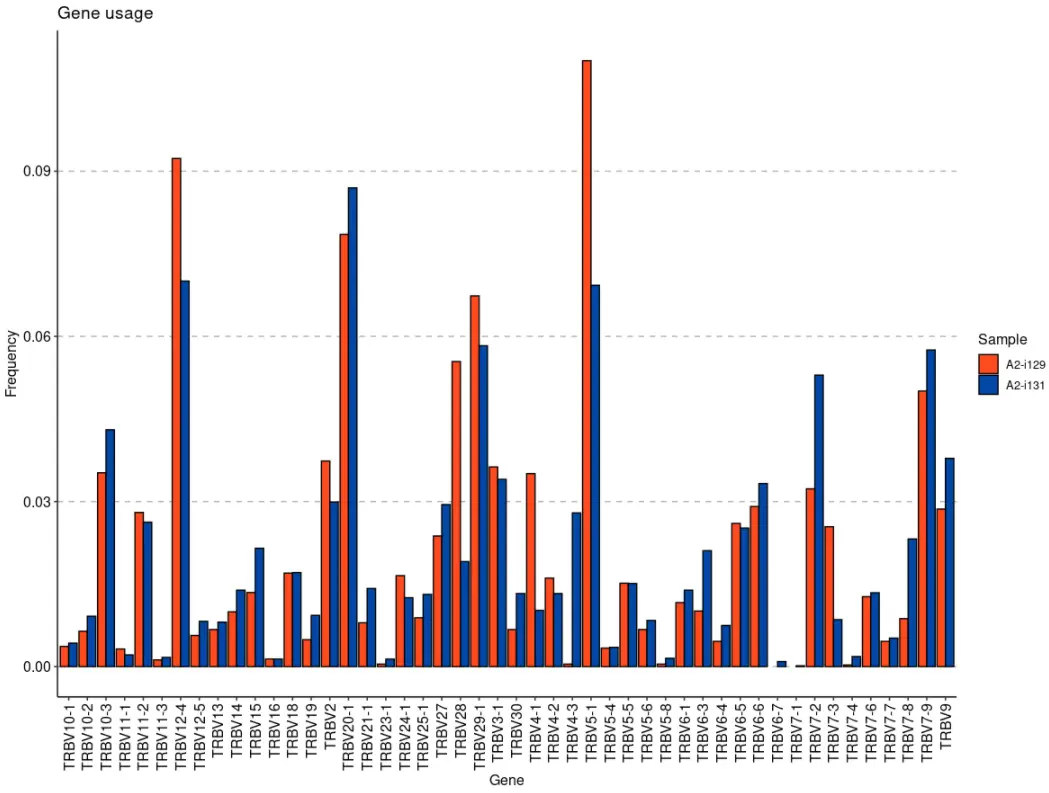
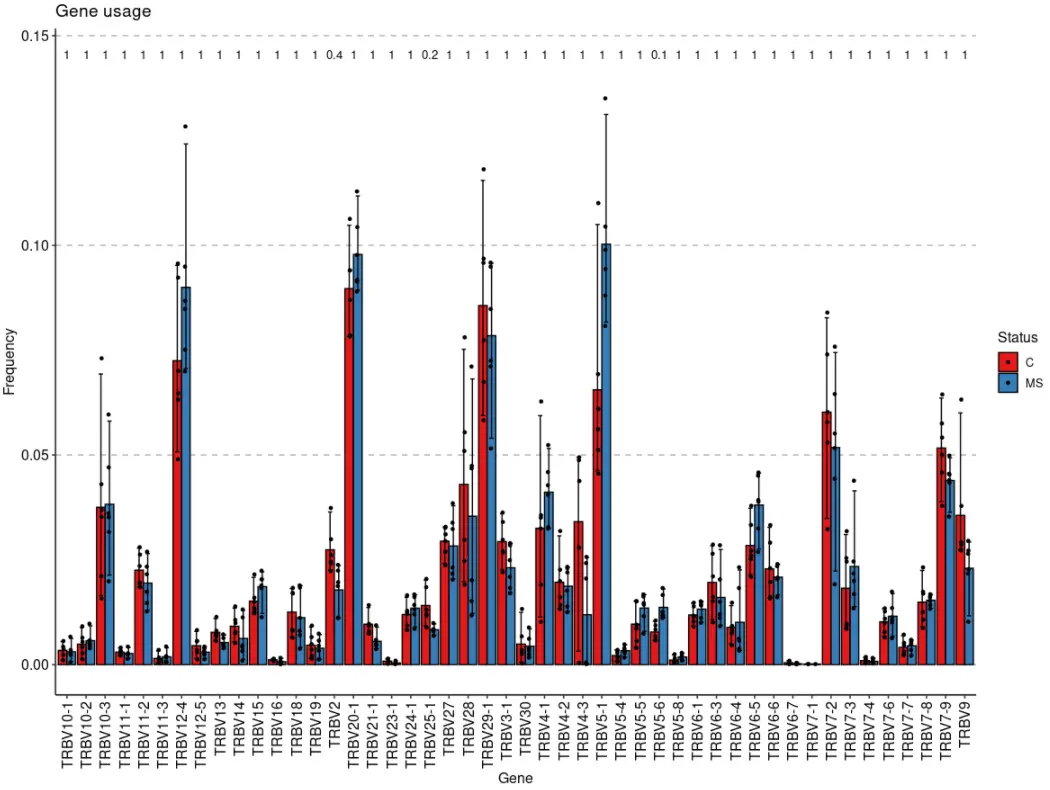
imm_gu <- geneUsage(immdata$data, "hs.trbv", .norm = T)vis(imm_gu, .by = "Status", .meta = immdata$meta)
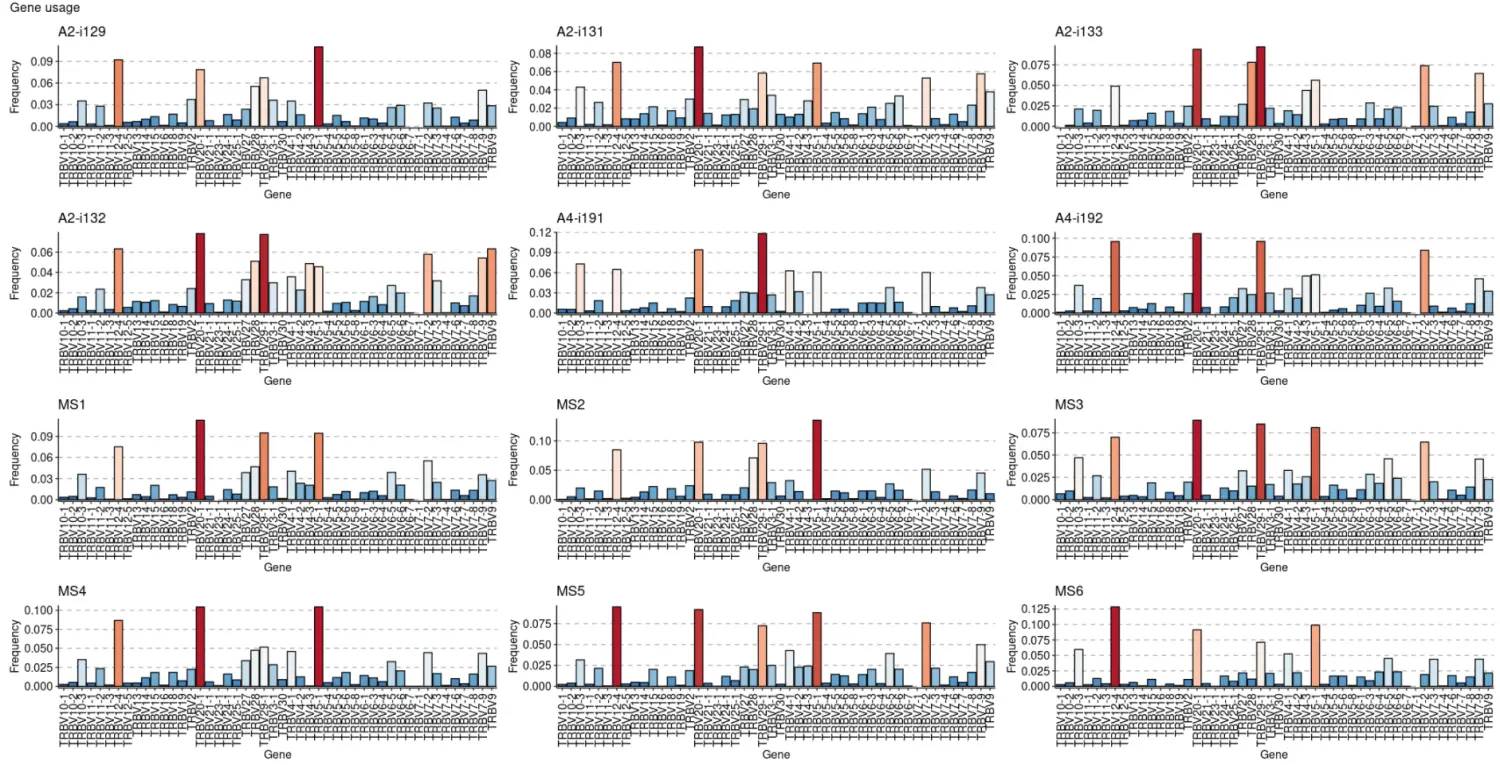
vis(imm_gu, .grid = T)
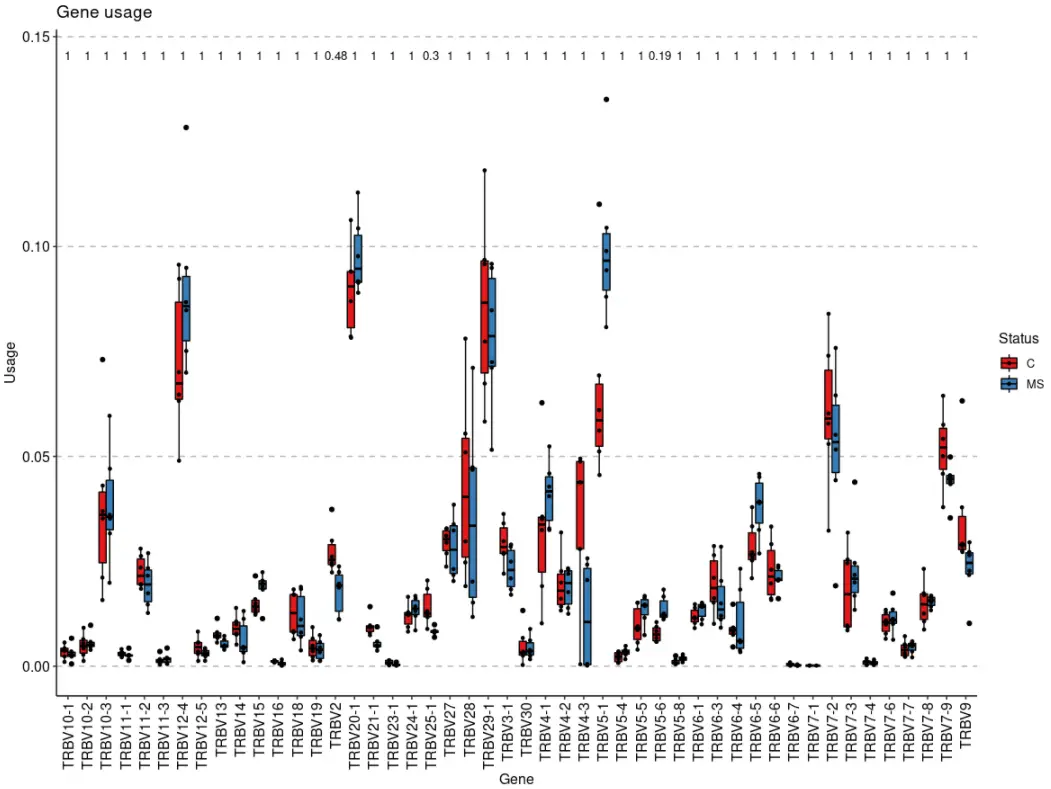
我们还可以使用箱线图进行可视化展示:
vis(imm_gu, .by = "Status", .meta = immdata$meta, .plot = "box")
基因片段名称的歧义
由于一些克隆型的基因序列比对的模糊性,geneUsage有以下选项来处理模糊数据:
.ambig = "inc"- 包含来自数据的模糊基因比对的所有可能组合。注意:ImmunoSEQ 格式使用非标准基因片段名称,因此最好将此参数值与 ImmunoSEQ 格式一起使用。默认情况下,此参数为 ON 以简化基因操作。如果是其他数据格式,请随意将其更改为"exc"。.ambig = "exc"- 过滤掉所有基因比对不明确的克隆型。.ambig = "wei"- 引入加权方法(1/n,如果存在n克隆型基因,则将相应基因的每个条目的频率除以 n)。.ambig = "maj"- 只选择第一个基因片段。
基因使用分析
在immunarch包中,我们可以使用geneUsageAnalysis函数进行基因使用分析。其中,.method参数可以控制如何预处理和分析数据。geneUsageAnalysis函数提供了以下预处理方法:
- “js” - Jensen-Shannon Divergence.
- “cor” - correlation.
- “cosine” - cosine similarity.
- “pca” - principal component analysis.
- “mds” - multi-dimensional scaling.
- “tsne” - t-Distributed Stochastic Neighbor Embedding.
以及一些常用分析的方法:
- “hclust” - 使用层次聚类对数据进行聚类。
- “kmeans” - 使用 K-means 对数据进行聚类。
- “dbscan” - 使用 DBSCAN 对数据进行聚类。
- “kruskall” - 在分成组的数据上分别计算每个基因的 Kruskall(未经预处理)。结果可与 Dunn 检验一起使用,以检测组间的显着差异。
我们可以在一行代码中调用多个方法,这可能是该包最强大的功能。例如,设置"js+hclust"参数首先计算 Jensen-Shannon离散度,然后在所得到的距离矩阵上进行层次聚类,而设置"anova"参数则可以在对所有基因进行分组后,分别对不同组基因进行方差分析:
imm_gu <- geneUsage(immdata$data, "hs.trbv", .norm = T)head(imm_gu)# A tibble: 6 x 13# Names `A2-i129` `A2-i131` `A2-i133` `A2-i132` `A4-i191` `A4-i192` MS1 MS2 MS3# <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>#1 TRBV… 0.00367 0.00427 NA 0.00234 0.00564 0.00103 3.52e-3 5.60e-4 0.00666#2 TRBV… 0.00643 0.00916 0.00125 0.00423 0.00544 0.00275 4.63e-3 4.90e-3 0.00975#3 TRBV… 0.0352 0.0430 0.0211 0.0158 0.0731 0.0369 3.61e-2 1.99e-2 0.0471#4 TRBV… 0.00321 0.00214 0.00407 0.00248 0.00330 0.00275 2.59e-3 1.40e-3 0.00248#5 TRBV… 0.0280 0.0262 0.0196 0.0235 0.0185 0.0194 1.74e-2 1.47e-2 0.0269#6 TRBV… 0.00122 0.00168 0.000782 0.00350 0.000389 0.00120 7.40e-4 1.82e-3 0.00201# … with 3 more variables: MS4 <dbl>, MS5 <dbl>, MS6 <dbl>imm_gu_js <- geneUsageAnalysis(imm_gu, .method = "js", .verbose = F)head(imm_gu_js)# A2-i129 A2-i131 A2-i133 A2-i132 A4-i191 A4-i192 MS1 MS2#A2-i129 NA 0.04541437 0.06045611 0.05765738 0.04840484 0.06990868 0.03736897 0.02463126#A2-i131 0.04541437 NA 0.04596597 0.04360022 0.06714223 0.04150936 0.05061679 0.06554060#A2-i133 0.06045611 0.04596597 NA 0.02509983 0.07427902 0.03294719 0.03674209 0.05589469#A2-i132 0.05765738 0.04360022 0.02509983 NA 0.07003044 0.03385875 0.03528860 0.07670967#A4-i191 0.04840484 0.06714223 0.07427902 0.07003044 NA 0.04410131 0.03682708 0.05610528#A4-i192 0.06990868 0.04150936 0.03294719 0.03385875 0.04410131 NA 0.03215804 0.07190250# MS3 MS4 MS5 MS6#A2-i129 0.04869536 0.01342004 0.04024095 0.04974138#A2-i131 0.03022664 0.04277057 0.03618735 0.08821618#A2-i133 0.04595669 0.06008131 0.04202235 0.11655132#A2-i132 0.04296950 0.05906025 0.03606000 0.09669160#A4-i191 0.03905298 0.04373909 0.04553688 0.05604907#A4-i192 0.02753266 0.06296453 0.02463658 0.08874825imm_gu_cor <- geneUsageAnalysis(imm_gu, .method = "cor", .verbose = F)head(imm_gu_cor)# A2-i129 A2-i131 A2-i133 A2-i132 A4-i191 A4-i192 MS1 MS2#A2-i129 NA 0.8794770 0.7952606 0.7997958 0.8144834 0.7972405 0.9098790 0.9531370#A2-i131 0.8794770 NA 0.8464620 0.8572172 0.8246651 0.9098564 0.8895600 0.8446886#A2-i133 0.7952606 0.8464620 NA 0.9234689 0.8084918 0.8912909 0.8858687 0.8448900#A2-i132 0.7997958 0.8572172 0.9234689 NA 0.8051677 0.9071005 0.8773288 0.7787694#A4-i191 0.8144834 0.8246651 0.8084918 0.8051677 NA 0.8882142 0.9135747 0.8202941#A4-i192 0.7972405 0.9098564 0.8912909 0.9071005 0.8882142 NA 0.9114279 0.8066513# MS3 MS4 MS5 MS6#A2-i129 0.8679673 0.9611283 0.9024905 0.8935665#A2-i131 0.9257652 0.8913960 0.9267036 0.8111305#A2-i133 0.8529126 0.7942104 0.8431999 0.6459174#A2-i132 0.8483939 0.7979125 0.8727292 0.7110267#A4-i191 0.9177682 0.8326876 0.8761637 0.8227976#A4-i192 0.9368741 0.8307886 0.9393554 0.7880824p1 <- vis(imm_gu_js, .title = "Gene usage JS-divergence", .leg.title = "JS", .text.size = 1.5)p2 <- vis(imm_gu_cor, .title = "Gene usage correlation", .leg.title = "Cor", .text.size = 1.5)p1 + p2
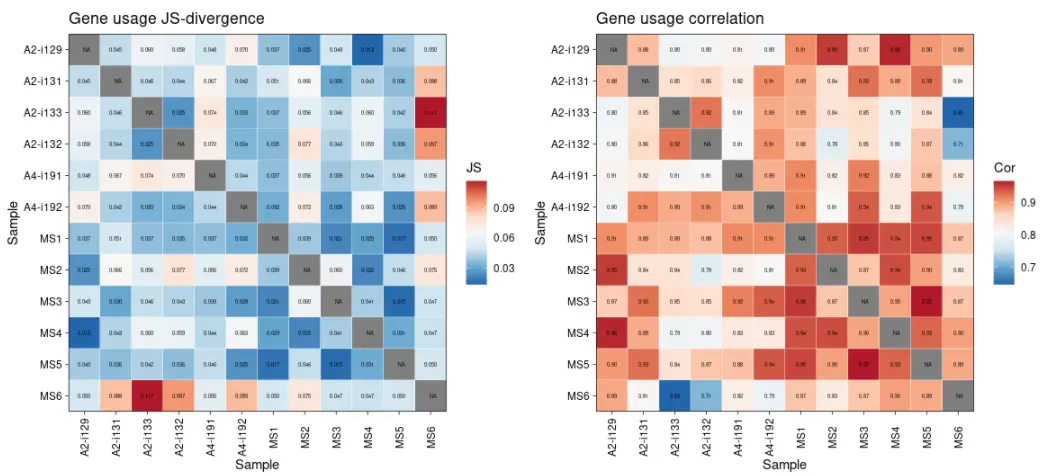
现在,让我们可视化预处理和分析后的输出结果:
imm_gu_js [ is.na ( imm_gu_js )] <- 0vis ( geneUsageAnalysis ( imm_gu , "cosine+hclust" , .verbose = F ))
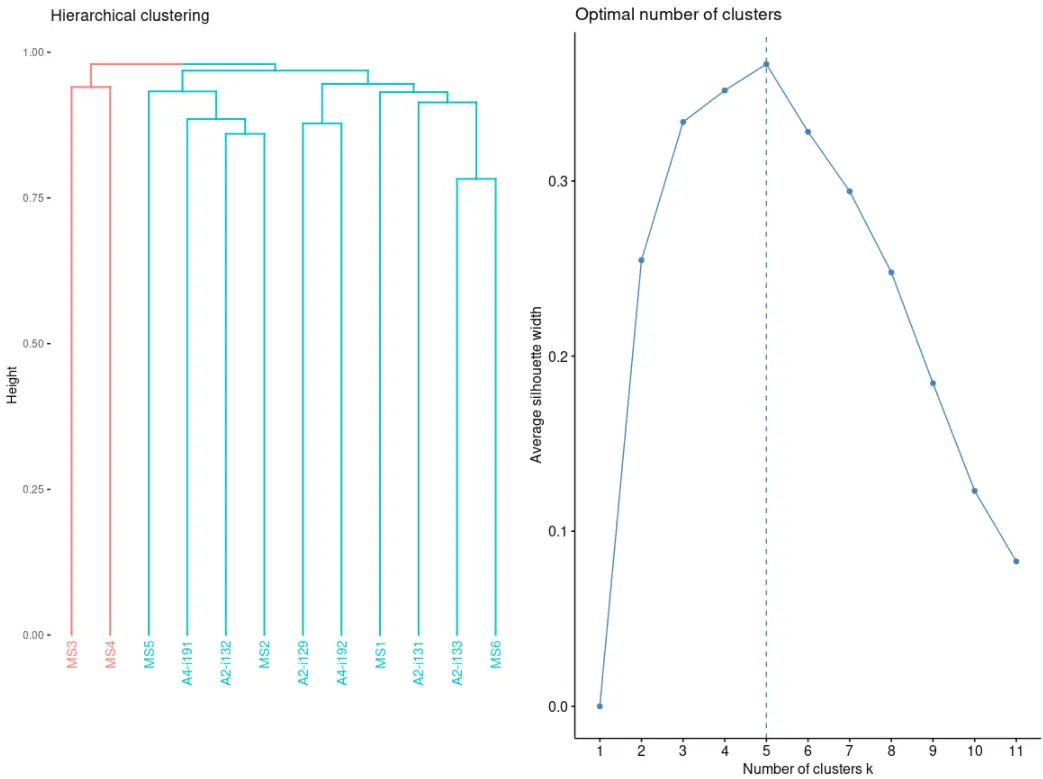
vis(geneUsageAnalysis(imm_gu, "js+dbscan", .verbose = F))
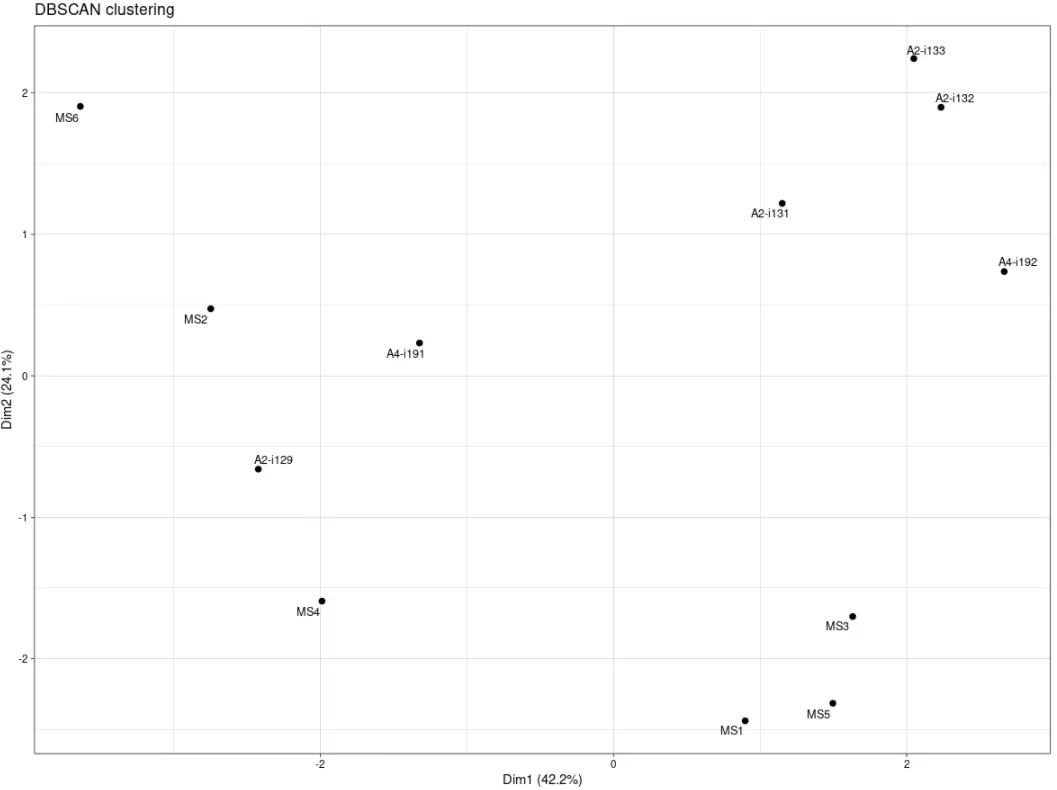
最重要的是,我们还可以进行聚类分群:
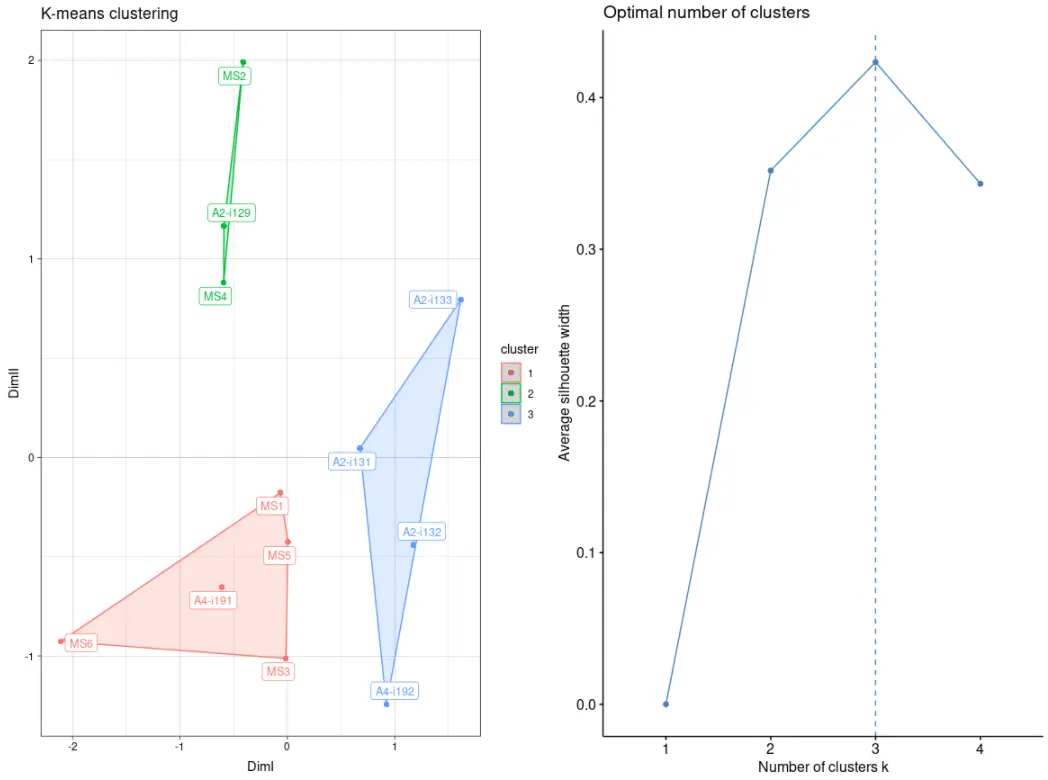
imm_cl_pca <- geneUsageAnalysis(imm_gu, "js+pca+kmeans", .verbose = F)imm_cl_mds <- geneUsageAnalysis(imm_gu, "js+mds+kmeans", .verbose = F)imm_cl_tsne <- geneUsageAnalysis(imm_gu, "js+tsne+kmeans", .perp = .01, .verbose = F)## Perplexity should be lower than K!
p1 <- vis(imm_cl_pca, .plot = "clust")p2 <- vis(imm_cl_mds, .plot = "clust")p3 <- vis(imm_cl_tsne, .plot = "clust")p1 + p2 + p3
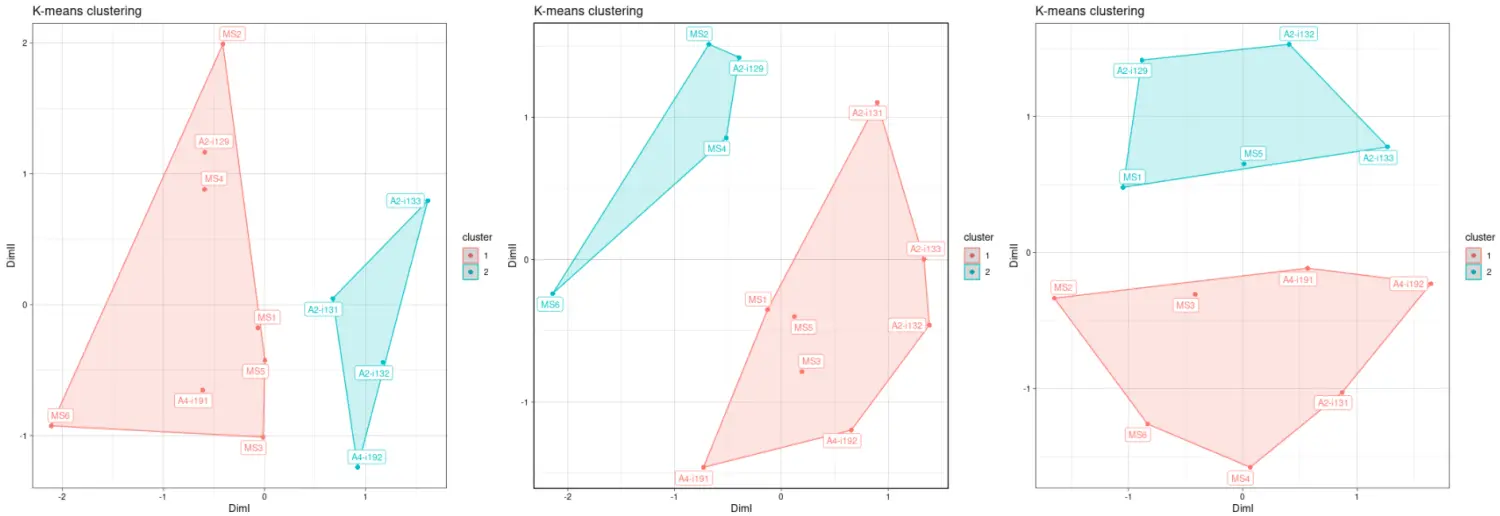
您也可以更改设置聚类群的数目:
imm_cl_pca2 <- geneUsageAnalysis(imm_gu, "js+pca+kmeans", .k = 3, .verbose = F)vis(imm_cl_pca2)
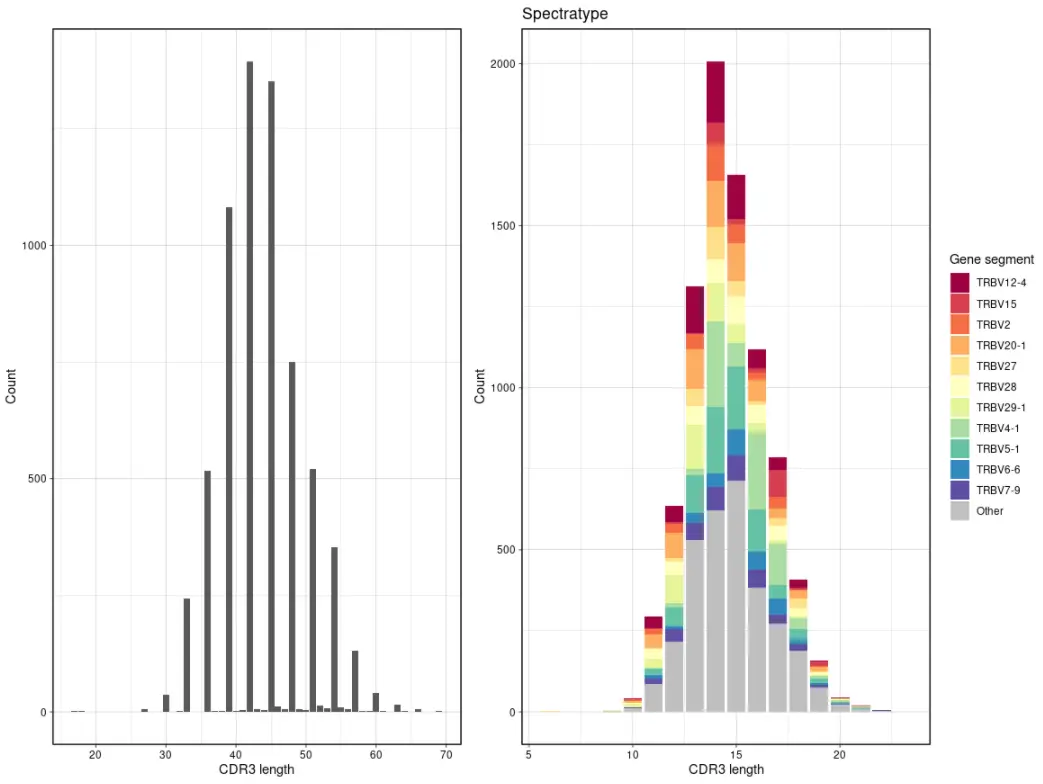
Spectratyping
Spectratype函数是一种计算每个序列长度的基因分布的有用方法。其中,参数.quant可以控制用于计算基因比例的数量 - 可以按克隆型 (id) 或按每个克隆型的克隆数 (count)。参数.col则控制选择哪一列,例如,“nt”表示仅计算CDR3区域核苷酸序列的长度(不按基因片段分组),“aa+v”表示计算CDR3区域氨基酸序列的长度(按 V 基因片段分组)。
p1 <- vis(spectratype(immdata$data[[1]], .quant = "id", .col = "nt"))p2 <- vis(spectratype(immdata$data[[1]], .quant = "count", .col = "aa+v"))p1 + p2
参考来源:https://immunarch.com/articles/web_only/v5_gene_usage.html

若有收获,就点个赞吧
0 人点赞
