- 10X Adult Mouse Brain
- 分析内容
- Step 0. Data download
- Step 1. Barcode selection
- Step 2. Add cell-by-bin matrix
- Step 3. Matrix binarization
- Step 4. Bin filtering
- Step 5. Dimensionality reduction
- Step 6. Determine significant components
- Step 7. Graph-based clustering
- Step 8. Visualization
- Step 9. Gene based annotation
- Step 10. Heretical clustering
- Step 11. Identify peaks
- Step 12. Create a cell-by-peak matrix
- Step 13. Add cell-by-peak matrix
- Step 14. Identify differentially accessible regions
- Step 15. Motif analysis identifies master regulators
- Step 16. GREAT analysis
10X Adult Mouse Brain
在本示例中,我们将分析来自10X genomics平台测序产生的5000个成年小鼠大脑细胞的数据集。该示例数据可以从以下网址进行下载:http://renlab.sdsc.edu/r3fang/share/github/Mouse_Brain_10X/
分析内容
- Step 0. Data download
- Step 1. Barcode selection
- Step 2. Add cell-by-bin matrix
- Step 3. Matrix binarization
- Step 4. Bin filtering
- Step 5. Dimensionality reduction
- Step 6. Determine significant components
- Step 7. Graph-based clustering
- Step 8. Visualization
- Step 9. Gene based annotation
- Step 10. Heretical clustering
- Step 11. Identify peak
- Step 12. Create a cell-by-peak matrix
- Step 13. Add cell-by-peak matrix
- Step 14. Identify differentially accessible regions
- Step 15. Motif analysis
- Step 16. GREAT analysis
Step 0. Data download
在本示例中,我们将直接下载所需的snap文件,该文件中已经包含了cell-by-bin/cell-by-peak matrix的计数矩阵。
wget http://renlab.sdsc.edu/r3fang/share/github/Mouse_Brain_10X/atac_v1_adult_brain_fresh_5k.snapwget http://renlab.sdsc.edu/r3fang/share/github/Mouse_Brain_10X/atac_v1_adult_brain_fresh_5k_singlecell.csv
Step 1. Barcode selection
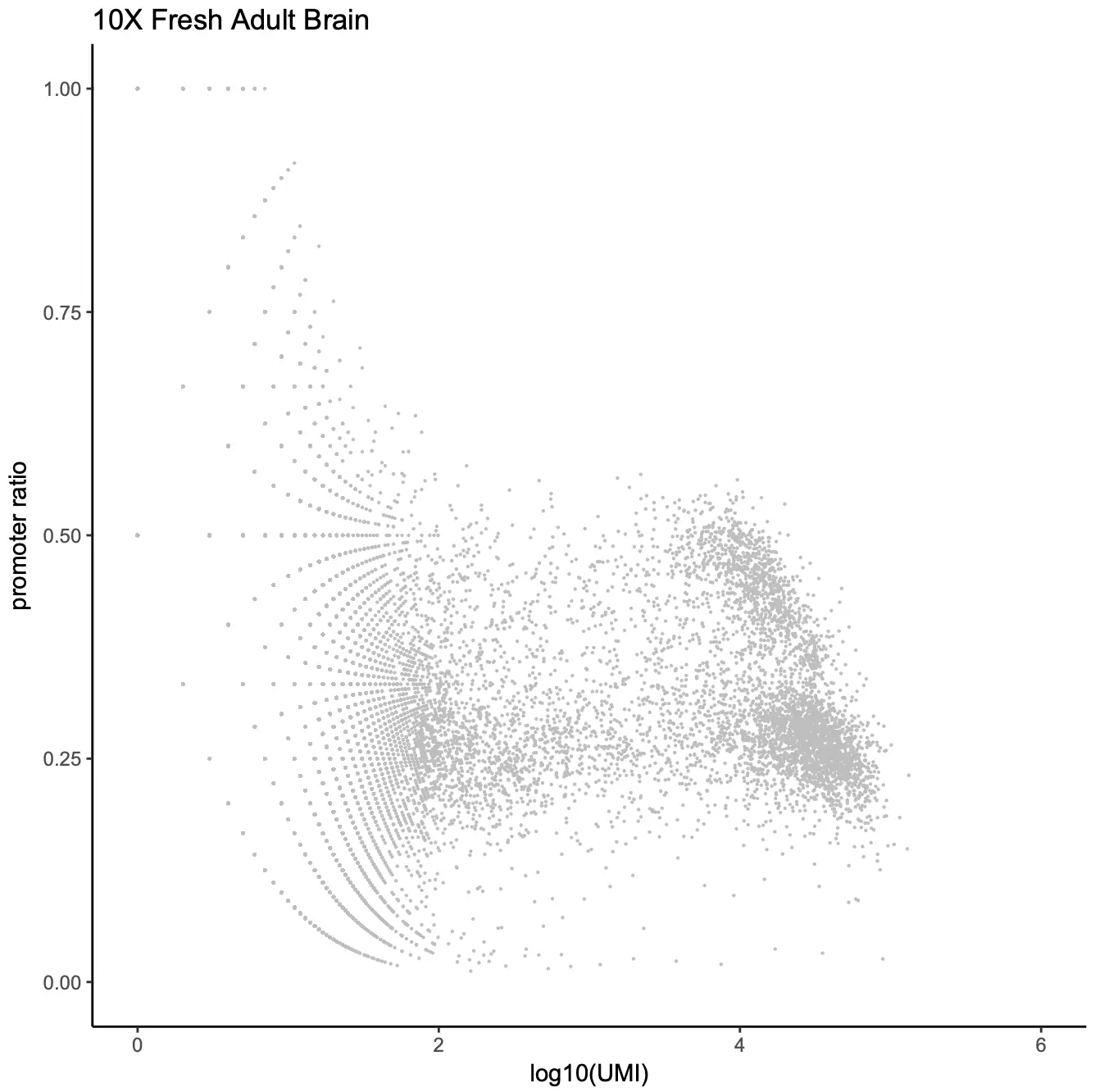
首先,我们对数据进行初步的过滤,基于以下标准选择出高质量barcodes的细胞: 1) number of unique fragments; 2) fragments in promoter ratio;
# 加载SnapATAC包library(SnapATAC);# 使用createSnap函数构建snap对象x.sp = createSnap(file="atac_v1_adult_brain_fresh_5k.snap",sample="atac_v1_adult_brain_fresh_5k",num.cores=1);# 读取barcode信息barcodes = read.csv("atac_v1_adult_brain_fresh_5k_singlecell.csv",head=TRUE);barcodes = barcodes[2:nrow(barcodes),];# 计算比对到promoter区域的比率promoter_ratio = (barcodes$promoter_region_fragments+1) / (barcodes$passed_filters + 1);UMI = log(barcodes$passed_filters+1, 10);data = data.frame(UMI=UMI, promoter_ratio=promoter_ratio);barcodes$promoter_ratio = promoter_ratio;library(viridisLite);library(ggplot2);p1 = ggplot(data,aes(x= UMI, y= promoter_ratio)) +geom_point(size=0.1, col="grey") +theme_classic() +ggtitle("10X Fresh Adult Brain") +ylim(0, 1) + xlim(0, 6) +labs(x = "log10(UMI)", y="promoter ratio")p1# 根据条件筛选barcodebarcodes.sel = barcodes[which(UMI >= 3 & UMI <= 5 & promoter_ratio >= 0.15 & promoter_ratio <= 0.6),];rownames(barcodes.sel) = barcodes.sel$barcode;x.sp = x.sp[which(x.sp@barcode %in% barcodes.sel$barcode),];x.sp@metaData = barcodes.sel[x.sp@barcode,];x.sp## number of barcodes: 4100## number of bins: 0## number of genes: 0## number of peaks: 0## number of motifs: 0
Step 2. Add cell-by-bin matrix
接下来,我们将5kb分辨率的cell-by-bin计数矩阵添加到snap对象中。addBmatToSnap函数将自动读取cell-by-bin计数矩阵,并将其添加到snap对象的bmat slot 中。
# show what bin sizes exist in atac_v1_adult_brain_fresh_5k.snap file
# 使用showBinSizes函数查看snap文件中的bin size信息
showBinSizes("atac_v1_adult_brain_fresh_5k.snap");
[1] 1000 5000 10000
# 使用addBmatToSnap函数添加cell-by-bin计数矩阵
x.sp = addBmatToSnap(x.sp, bin.size=5000, num.cores=1);
Step 3. Matrix binarization
接下来,我们将cell-by-bin的计数矩阵转换为二进制矩阵。计数矩阵中的某些items具有异常高的覆盖率,这可能是由比对错误造成的。因此,我们会将计数矩阵中覆盖率最高的0.1%的items进行删除,然后将其余的非零的items转换为1。
# 使用makeBinary函数将计数矩阵转换为二进制矩阵
x.sp = makeBinary(x.sp, mat="bmat");
Step 4. Bin filtering
首先,我们将过滤掉与ENCODE blacklist区域重叠的bins,避免潜在的人为因素产生的误差。
system("wget http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/mm10-mouse/mm10.blacklist.bed.gz");
library(GenomicRanges);
black_list = read.table("mm10.blacklist.bed.gz");
black_list.gr = GRanges(
black_list[,1],
IRanges(black_list[,2], black_list[,3])
);
idy = queryHits(findOverlaps(x.sp@feature, black_list.gr));
if(length(idy) > 0){x.sp = x.sp[,-idy, mat="bmat"]};
x.sp
## number of barcodes: 4100
## number of bins: 546103
## number of genes: 0
## number of peaks: 0
## number of motifs: 0
接下来,我们将移除那些不需要的染色体上的信息。
chr.exclude = seqlevels(x.sp@feature)[grep("random|chrM", seqlevels(x.sp@feature))];
idy = grep(paste(chr.exclude, collapse="|"), x.sp@feature);
if(length(idy) > 0){x.sp = x.sp[,-idy, mat="bmat"]};
x.sp
## number of barcodes: 4100
## number of bins: 545183
## number of genes: 0
## number of peaks: 0
## number of motifs: 0
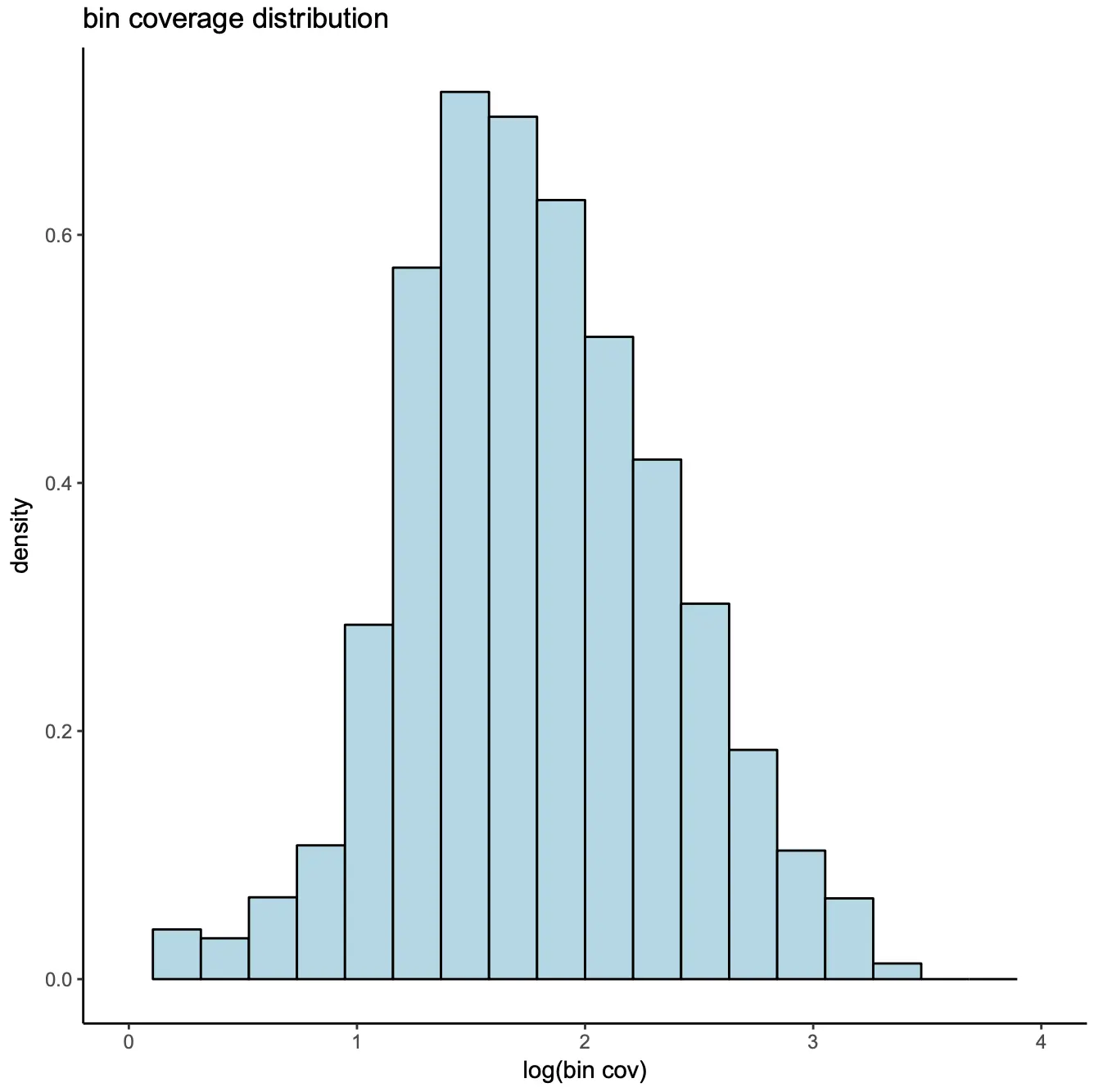
最后,bin的覆盖率大致服从对数正态分布(log normal distribution),我们会将与invariant features(如管家基因的启动子)区域重叠的前5%的bins进行删除。
bin.cov = log10(Matrix::colSums(x.sp@bmat)+1);
hist(
bin.cov[bin.cov > 0],
xlab="log10(bin cov)",
main="log10(Bin Cov)",
col="lightblue",
xlim=c(0, 5)
);
bin.cutoff = quantile(bin.cov[bin.cov > 0], 0.95);
idy = which(bin.cov <= bin.cutoff & bin.cov > 0);
x.sp = x.sp[, idy, mat="bmat"];
x.sp
## number of barcodes: 4100
## number of bins: 474624
## number of genes: 0
## number of peaks: 0
## number of motifs: 0
Step 5. Dimensionality reduction
使用diffusion maps的方法进行数据降维
x.sp = runDiffusionMaps(
obj=x.sp,
input.mat="bmat",
num.eigs=50
);
Step 6. Determine significant components
接下来,我们基于数据降维的结果确定用于下游分析的维数。我们绘制不同维数之间的配对散点图,并选择散点图开始看起来零散的维数。在下面的示例中,我们选择了前20个维度。
plotDimReductPW(
obj=x.sp,
eigs.dims=1:50,
point.size=0.3,
point.color="grey",
point.shape=19,
point.alpha=0.6,
down.sample=5000,
pdf.file.name=NULL,
pdf.height=7,
pdf.width=7
);

Step 7. Graph-based clustering
选择好有效的降维维度后,我们基于它们来构造一个K近邻(KNN)的聚类图(K =15)。在该图中,每个点代表一个细胞,并根据欧氏距离确定每个细胞的k近邻个点。
x.sp = runKNN(
obj=x.sp,
eigs.dims=1:20,
k=15
);
x.sp=runCluster(
obj=x.sp,
tmp.folder=tempdir(),
louvain.lib="R-igraph",
seed.use=10
);
x.sp@metaData$cluster = x.sp@cluster;
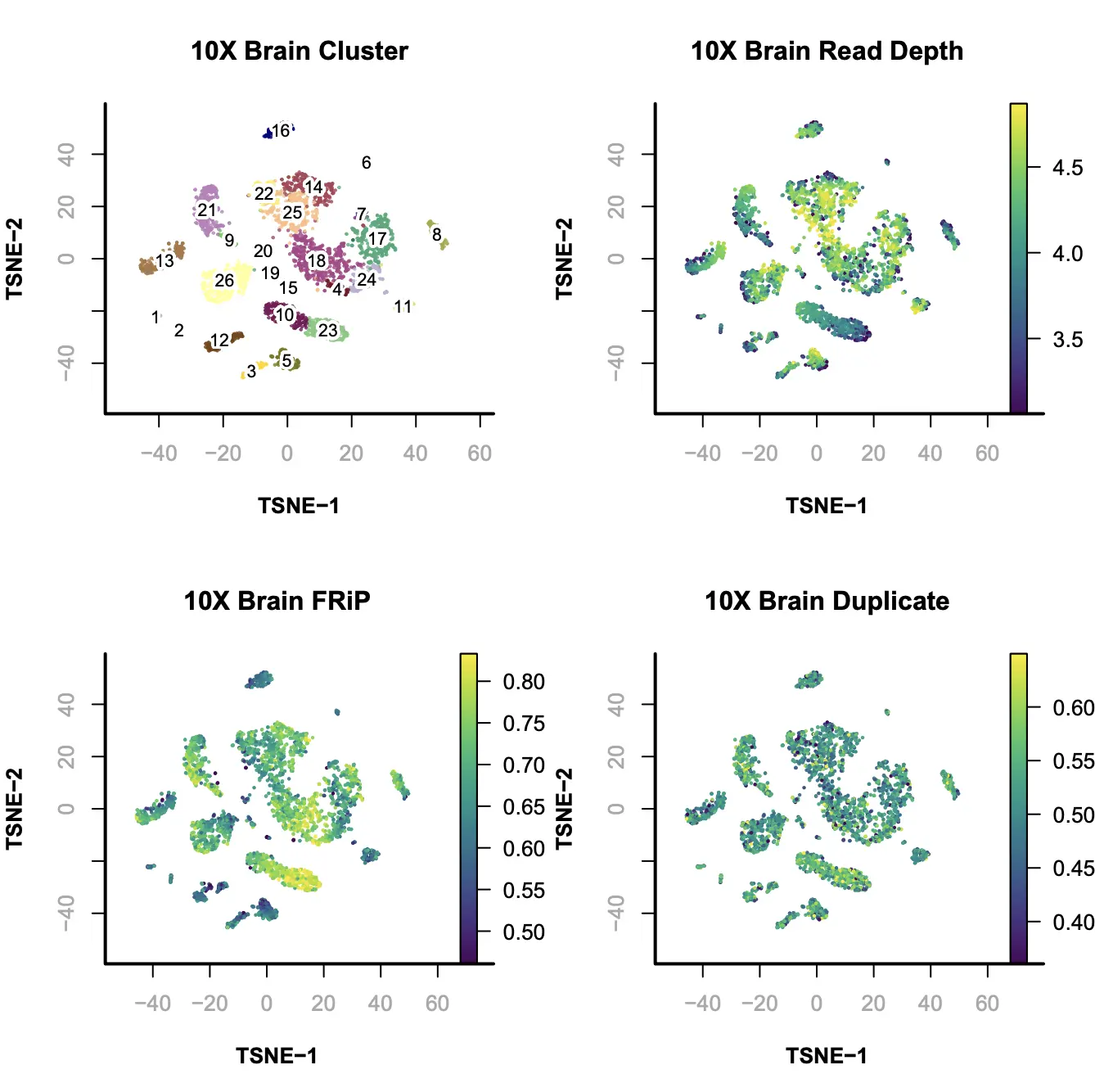
Step 8. Visualization
SnapATAC可以使用tSNE(FI-tsne)或UMAP的方法对降维聚类后的结果进行可视化展示。在此例中,我们计算t-SNE embedding,使用tSNE方法进行可视化展示。同时,我们还将测序深度或其他偏差投射到t-SNE embedding上。
x.sp = runViz(
obj=x.sp,
tmp.folder=tempdir(),
dims=2,
eigs.dims=1:20,
method="Rtsne",
seed.use=10
);
par(mfrow = c(2, 2));
plotViz(
obj=x.sp,
method="tsne",
main="10X Brain Cluster",
point.color=x.sp@cluster,
point.size=1,
point.shape=19,
point.alpha=0.8,
text.add=TRUE,
text.size=1.5,
text.color="black",
text.halo.add=TRUE,
text.halo.color="white",
text.halo.width=0.2,
down.sample=10000,
legend.add=FALSE
);
plotFeatureSingle(
obj=x.sp,
feature.value=log(x.sp@metaData[,"passed_filters"]+1,10),
method="tsne",
main="10X Brain Read Depth",
point.size=0.2,
point.shape=19,
down.sample=10000,
quantiles=c(0.01, 0.99)
);
plotFeatureSingle(
obj=x.sp,
feature.value=x.sp@metaData$peak_region_fragments / x.sp@metaData$passed_filters,
method="tsne",
main="10X Brain FRiP",
point.size=0.2,
point.shape=19,
down.sample=10000,
quantiles=c(0.01, 0.99) # remove outliers
);
plotFeatureSingle(
obj=x.sp,
feature.value=x.sp@metaData$duplicate / x.sp@metaData$total,
method="tsne",
main="10X Brain Duplicate",
point.size=0.2,
point.shape=19,
down.sample=10000,
quantiles=c(0.01, 0.99) # remove outliers
);

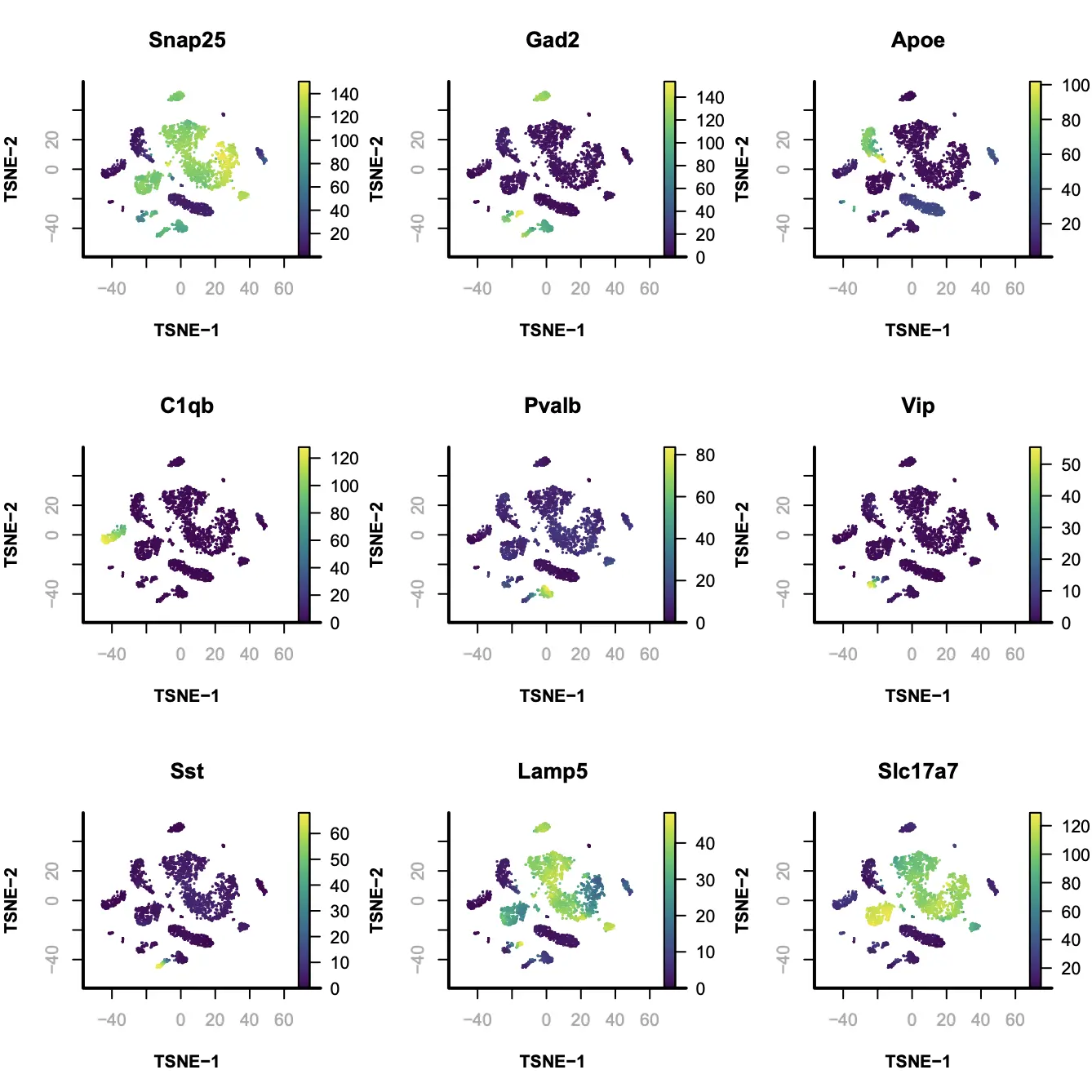
Step 9. Gene based annotation
为了帮助注释聚类后分群的细胞簇,SnapATAC接下来将创建cell-by-gene计数矩阵,并可视化marker基因的富集情况,根据marker基因的表达情况进行cluster的注释。
system("wget http://renlab.sdsc.edu/r3fang/share/github/Mouse_Brain_10X/gencode.vM16.gene.bed");
genes = read.table("gencode.vM16.gene.bed");
genes.gr = GRanges(genes[,1],
IRanges(genes[,2], genes[,3]), name=genes[,4]
);
marker.genes = c(
"Snap25", "Gad2", "Apoe",
"C1qb", "Pvalb", "Vip",
"Sst", "Lamp5", "Slc17a7"
);
genes.sel.gr <- genes.gr[which(genes.gr$name %in% marker.genes)];
# re-add the cell-by-bin matrix to the snap object;
x.sp = addBmatToSnap(x.sp);
x.sp = createGmatFromMat(
obj=x.sp,
input.mat="bmat",
genes=genes.sel.gr,
do.par=TRUE,
num.cores=10
);
# normalize the cell-by-gene matrix
x.sp = scaleCountMatrix(
obj=x.sp,
cov=x.sp@metaData$passed_filters + 1,
mat="gmat",
method = "RPM"
);
# smooth the cell-by-gene matrix
x.sp = runMagic(
obj=x.sp,
input.mat="gmat",
step.size=3
);
par(mfrow = c(3, 3));
for(i in 1:9){
plotFeatureSingle(
obj=x.sp,
feature.value=x.sp@gmat[, marker.genes[i]],
method="tsne",
main=marker.genes[i],
point.size=0.1,
point.shape=19,
down.sample=10000,
quantiles=c(0, 1)
)};
Step 10. Heretical clustering
接下来,我们将属于同一cluster的细胞汇集到一起,用以创建每个cluster的聚合信号(aggregate signal)。
# calculate the ensemble signals for each cluster
ensemble.ls = lapply(split(seq(length(x.sp@cluster)), x.sp@cluster), function(x){
SnapATAC::colMeans(x.sp[x,], mat="bmat");
})
# cluster using 1-cor as distance
hc = hclust(as.dist(1 - cor(t(do.call(rbind, ensemble.ls)))), method="ward.D2");
plotViz(
obj=x.sp,
method="tsne",
main="10X Brain Cluster",
point.color=x.sp@cluster,
point.size=1,
point.shape=19,
point.alpha=0.8,
text.add=TRUE,
text.size=1.5,
text.color="black",
text.halo.add=TRUE,
text.halo.color="white",
text.halo.width=0.2,
down.sample=10000,
legend.add=FALSE
);
plot(hc, hang=-1, xlab="");
在本示例中,cluster 20到25是兴奋性神经元细胞,cluster 19到5为抑制性神经元细胞,而其余的为非神经元细胞。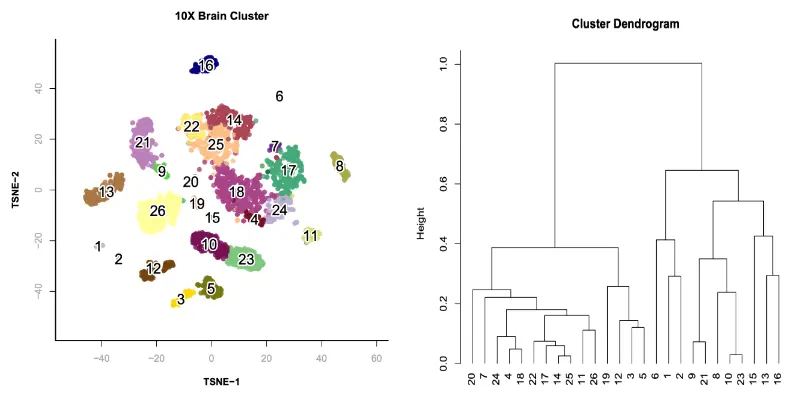
Step 11. Identify peaks
接下来,我们将每个cluster群的细胞信息聚合起来,创建一个用于peak calling和可视化的集成track。在该步骤中,将生成一个.narrowPeak的文件,其中包含识别出的所有peak的信息,和一个.bedGraph文件,可以用于可视化展示。为了获得最robust的结果,我们不建议对细胞数目小于100的cluster执行此步骤。
# 查看snaptools的安装路径
system("which snaptools")
/home/r3fang/anaconda2/bin/snaptools
# 查看macs2的安装路径
system("which macs2")
/home/r3fang/anaconda2/bin/macs2
# 调用macs2进行peak callling
runMACS(
obj=x.sp[which(x.sp@cluster==1),],
output.prefix="atac_v1_adult_brain_fresh_5k.1",
path.to.snaptools="/home/r3fang/anaconda2/bin/snaptools",
path.to.macs="/home/r3fang/anaconda2/bin/macs2",
gsize="mm",
buffer.size=500,
num.cores=5,
macs.options="--nomodel --shift 37 --ext 73 --qval 1e-2 -B --SPMR --call-summits",
tmp.folder=tempdir()
);
接下来,我们将提供一个简短的脚本,用于为所有的cluster进行批量操作此步骤。
# call peaks for all cluster with more than 100 cells
clusters.sel = names(table(x.sp@cluster))[which(table(x.sp@cluster) > 200)];
peaks.ls = mclapply(seq(clusters.sel), function(i){
print(clusters.sel[i]);
runMACS(
obj=x.sp[which(x.sp@cluster==clusters.sel[i]),],
output.prefix=paste0("atac_v1_adult_brain_fresh_5k.", gsub(" ", "_", clusters.sel)[i]),
path.to.snaptools="/home/r3fang/anaconda2/bin/snaptools",
path.to.macs="/home/r3fang/anaconda2/bin/macs2",
gsize="hs", # mm, hs, etc
buffer.size=500,
num.cores=1,
macs.options="--nomodel --shift 100 --ext 200 --qval 5e-2 -B --SPMR",
tmp.folder=tempdir()
);
}, mc.cores=5);
# assuming all .narrowPeak files in the current folder are generated from the clusters
peaks.names = system("ls | grep narrowPeak", intern=TRUE);
peak.gr.ls = lapply(peaks.names, function(x){
peak.df = read.table(x)
GRanges(peak.df[,1], IRanges(peak.df[,2], peak.df[,3]))
})
# 合并所有的peak信息
peak.gr = reduce(Reduce(c, peak.gr.ls));
peak.gr
## GRanges object with 242847 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 [3094889, 3095629] *
## [2] chr1 [3113499, 3114060] *
## [3] chr1 [3118103, 3118401] *
## [4] chr1 [3119689, 3120845] *
## [5] chr1 [3121534, 3121786] *
## ... ... ... ...
## [242843] chrY [90797373, 90798136] *
## [242844] chrY [90804709, 90805456] *
## [242845] chrY [90808580, 90808819] *
## [242846] chrY [90808850, 90809131] *
## [242847] chrY [90810817, 90811057] *
## -------
Step 12. Create a cell-by-peak matrix
接下来,我们基于合并后的peak信息作为参考,使用原始的snap文件创建一个 cell-by-peak的计数矩阵。
peaks.df = as.data.frame(peak.gr)[,1:3];
write.table(peaks.df,file = "peaks.combined.bed",append=FALSE,
quote= FALSE,sep="\t", eol = "\n", na = "NA", dec = ".",
row.names = FALSE, col.names = FALSE, qmethod = c("escape", "double"),
fileEncoding = "")
saveRDS(x.sp, file="atac_v1_adult_brain_fresh_5k.snap.rds");
我们使用snaptools创建cell-by-peak的计数矩阵,并将其添加到snap文件中,这一步可能需要一段时间。
snaptools snap-add-pmat \
--snap-file atac_v1_adult_brain_fresh_5k.snap \
--peak-file peaks.combined.bed
Step 13. Add cell-by-peak matrix
接下来,我们将计算好的cell-by-peak计数矩阵添加到现有的snap对象中。
x.sp = readRDS("atac_v1_adult_brain_fresh_5k.snap.rds");
x.sp = addPmatToSnap(x.sp);
x.sp = makeBinary(x.sp, mat="pmat");
x.sp
## number of barcodes: 4100
## number of bins: 546206
## number of genes: 16
## number of peaks: 242847
## number of motifs: 0
Step 14. Identify differentially accessible regions
SnapATAC通过差异分析来识别出不同cluster群中的差异可及性区域( differentially accessible regions,DARs)。默认情况下,它只寻找每个cluster群中的positive peaks(可以通过cluster.pos参数指定), 与一组阴性对照细胞相比。如果默认的cluster.neg=NULL, findDAR函数将寻找最接近positive细胞的一组作为背景细胞。
DARs = findDAR(
obj=x.sp,
input.mat="pmat",
cluster.pos=26,
cluster.neg.method="knn",
test.method="exactTest",
bcv=0.1, #0.4 for human, 0.1 for mouse
seed.use=10
);
DARs$FDR = p.adjust(DARs$PValue, method="BH");
idy = which(DARs$FDR < 5e-2 & DARs$logFC > 0);
par(mfrow = c(1, 2));
plot(DARs$logCPM, DARs$logFC,
pch=19, cex=0.1, col="grey",
ylab="logFC", xlab="logCPM",
main="Cluster 26"
);
points(DARs$logCPM[idy],
DARs$logFC[idy],
pch=19,
cex=0.5,
col="red"
);
abline(h = 0, lwd=1, lty=2);
covs = Matrix::rowSums(x.sp@pmat);
vals = Matrix::rowSums(x.sp@pmat[,idy]) / covs;
vals.zscore = (vals - mean(vals)) / sd(vals);
plotFeatureSingle(
obj=x.sp,
feature.value=vals.zscore,
method="tsne",
main="Cluster 26",
point.size=0.1,
point.shape=19,
down.sample=5000,
quantiles=c(0.01, 0.99)
);
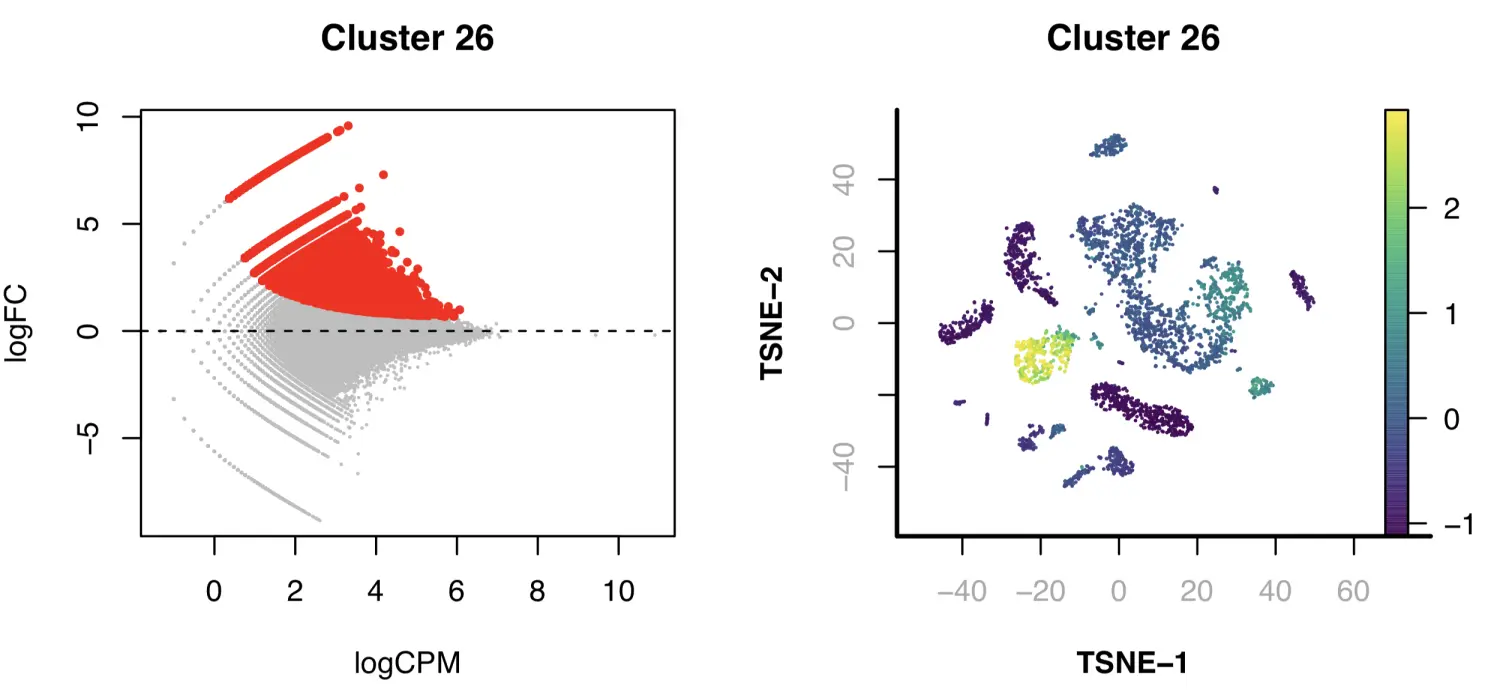
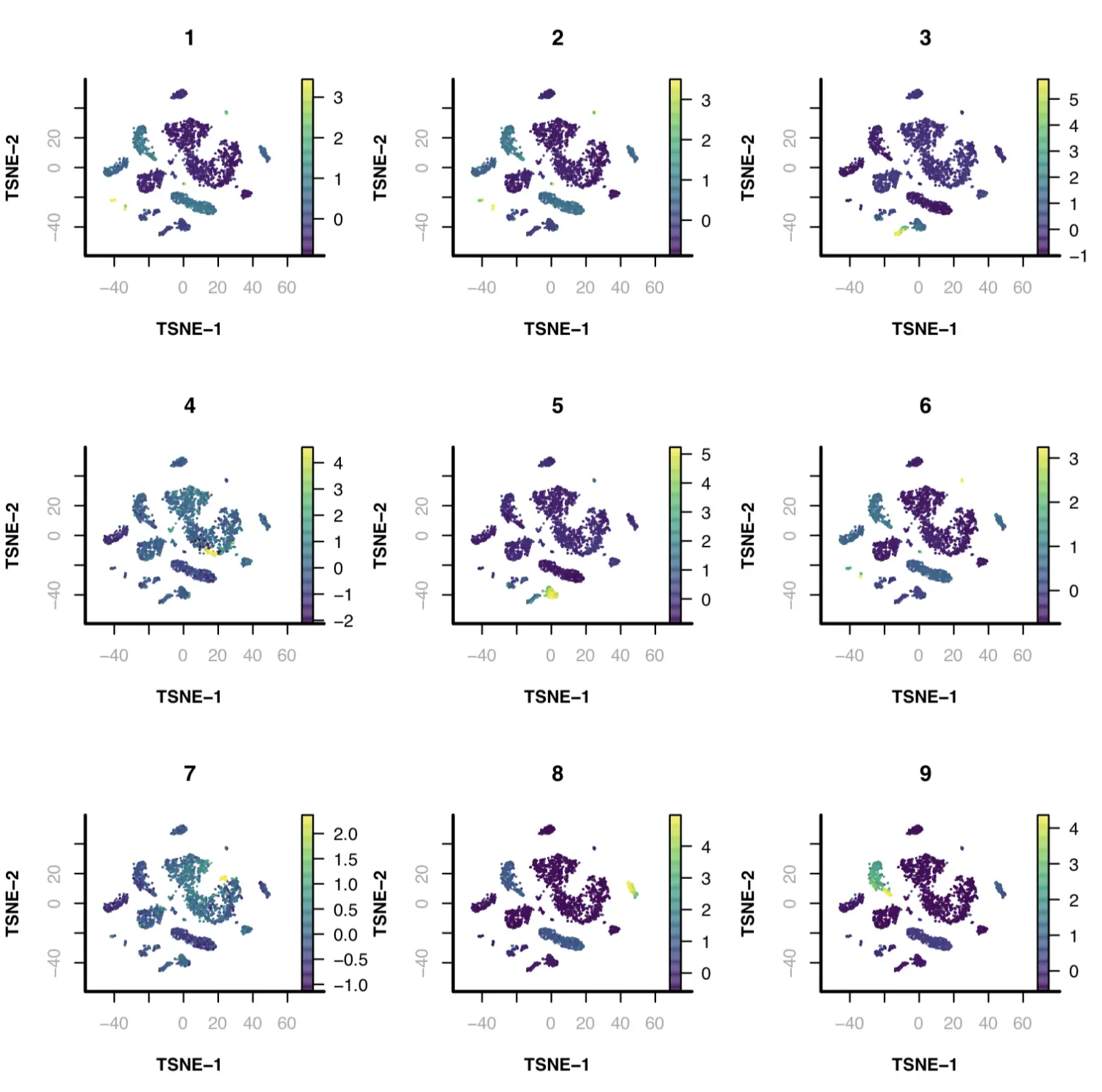
接下来,我们识别出每个cluster群中的DARs。对于缺乏揭示DARs的静态能力(static power)的簇,特别是比较小的簇,我们根据peak的富集程度对其进行排序,并使用top 2000个peak用于motif discovery的代表性峰。
idy.ls = lapply(levels(x.sp@cluster), function(cluster_i){
DARs = findDAR(
obj=x.sp,
input.mat="pmat",
cluster.pos=cluster_i,
cluster.neg=NULL,
cluster.neg.method="knn",
bcv=0.1,
test.method="exactTest",
seed.use=10
);
DARs$FDR = p.adjust(DARs$PValue, method="BH");
idy = which(DARs$FDR < 5e-2 & DARs$logFC > 0);
if((x=length(idy)) < 2000L){
PValues = DARs$PValue;
PValues[DARs$logFC < 0] = 1;
idy = order(PValues, decreasing=FALSE)[1:2000];
rm(PValues); # free memory
}
idy
})
names(idy.ls) = levels(x.sp@cluster);
par(mfrow = c(3, 3));
for(cluster_i in levels(x.sp@cluster)){
print(cluster_i)
idy = idy.ls[[cluster_i]];
vals = Matrix::rowSums(x.sp@pmat[,idy]) / covs;
vals.zscore = (vals - mean(vals)) / sd(vals);
plotFeatureSingle(
obj=x.sp,
feature.value=vals.zscore,
method="tsne",
main=cluster_i,
point.size=0.1,
point.shape=19,
down.sample=5000,
quantiles=c(0.01, 0.99)
);
}
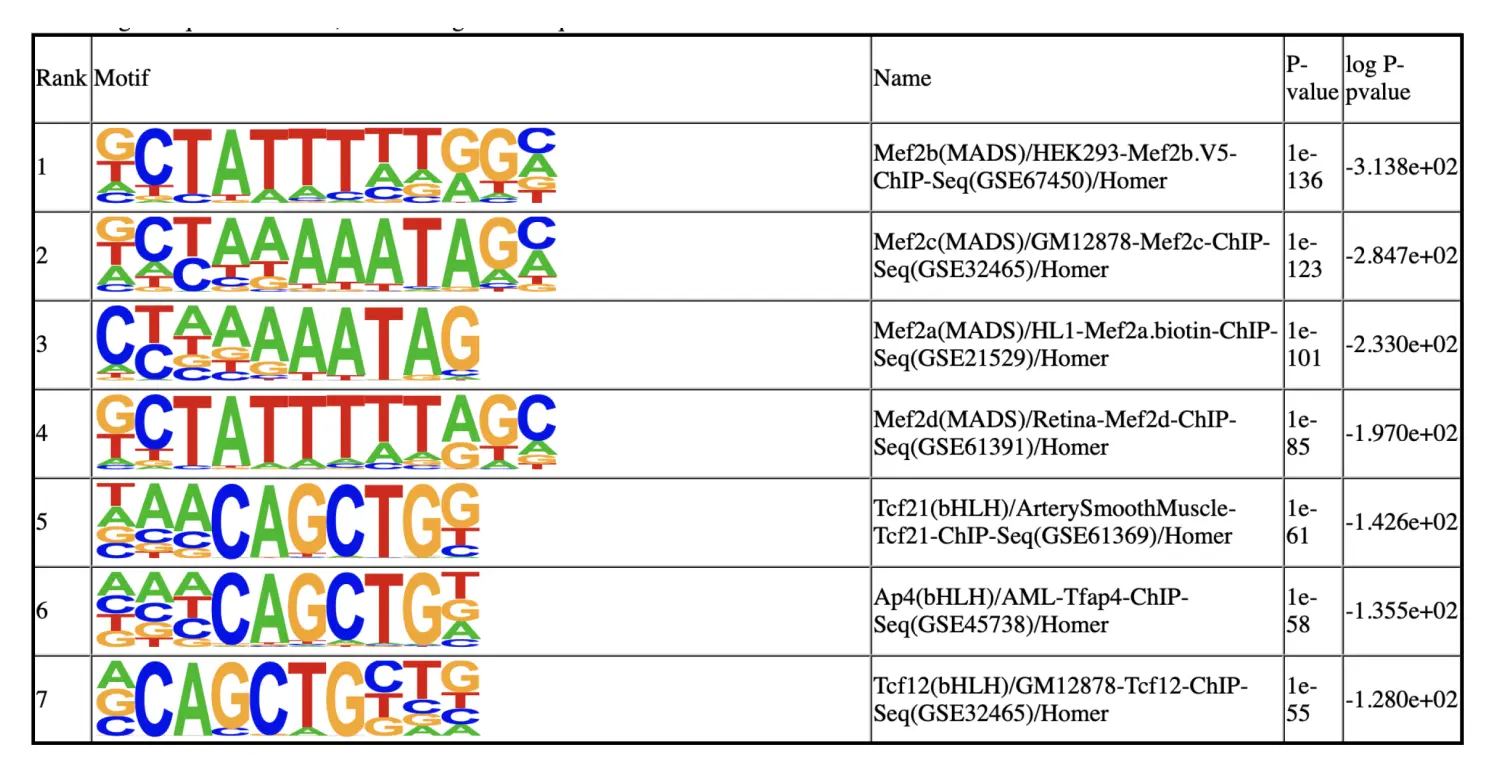
Step 15. Motif analysis identifies master regulators
SnapATAC可以调用Homer来鉴定识别出的差异可及性区域(DARs)中富集的master regulators。运行完runHomer函数后,会在./homer/C5文件夹中生成一个homer motif的报告knownResults.html。我们需要预先安装好Homer程序。
system("which findMotifsGenome.pl");
/projects/ps-renlab/r3fang/public_html/softwares/homer/bin/findMotifsGenome.pl
motifs = runHomer(
x.sp[,idy.ls[["5"]],"pmat"],
mat = "pmat",
path.to.homer = "/projects/ps-renlab/r3fang/public_html/softwares/homer/bin/findMotifsGenome.pl",
result.dir = "./homer/C5",
num.cores=5,
genome = 'mm10',
motif.length = 10,
scan.size = 300,
optimize.count = 2,
background = 'automatic',
local.background = FALSE,
only.known = TRUE,
only.denovo = FALSE,
fdr.num = 5,
cache = 100,
overwrite = TRUE,
keep.minimal = FALSE
);
SnapATAC还可以调用chromVAR(Schep等)来进行motif的可变性分析。
library(chromVAR);
library(motifmatchr);
library(SummarizedExperiment);
library(BSgenome.Mmusculus.UCSC.mm10);
x.sp = makeBinary(x.sp, "pmat");
x.sp@mmat = runChromVAR(
obj=x.sp,
input.mat="pmat",
genome=BSgenome.Mmusculus.UCSC.mm10,
min.count=10,
species="Homo sapiens"
);
motif_i = "MA0497.1_MEF2C";
dat = data.frame(x=x.sp@metaData[,"cluster"], y=x.sp@mmat[,motif_i]);
p1 <- ggplot(dat, aes(x=x, y=y, fill=x)) +
theme_classic() +
geom_violin() +
xlab("cluster") +
ylab("motif enrichment") +
ggtitle(motif_i) +
theme(
plot.margin = margin(5,1,5,1, "cm"),
axis.text.x = element_text(angle = 90, hjust = 1),
axis.ticks.x=element_blank(),
legend.position = "none"
);
motif_i = "MA0660.1_MEF2B";
dat = data.frame(x=x.sp@metaData[,"cluster"], y=x.sp@mmat[,motif_i]);
p2 <- ggplot(dat, aes(x=x, y=y, fill=x)) +
theme_classic() +
geom_violin() +
xlab("cluster") +
ylab("motif enrichment") +
ggtitle(motif_i) +
theme(
plot.margin = margin(5,1,5,1, "cm"),
axis.text.x = element_text(angle = 90, hjust = 1),
axis.ticks.x=element_blank(),
legend.position = "none"
);
p1
p2
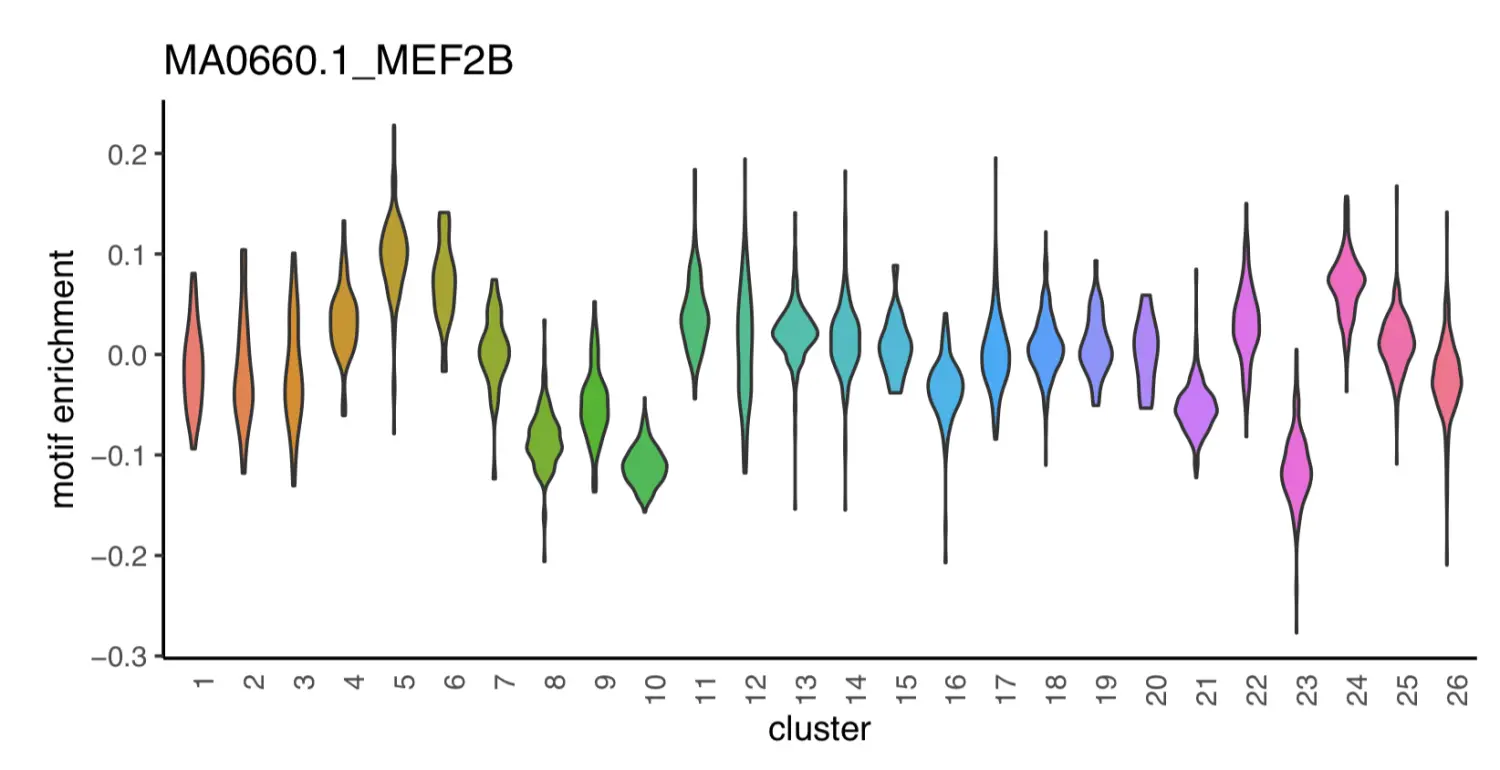
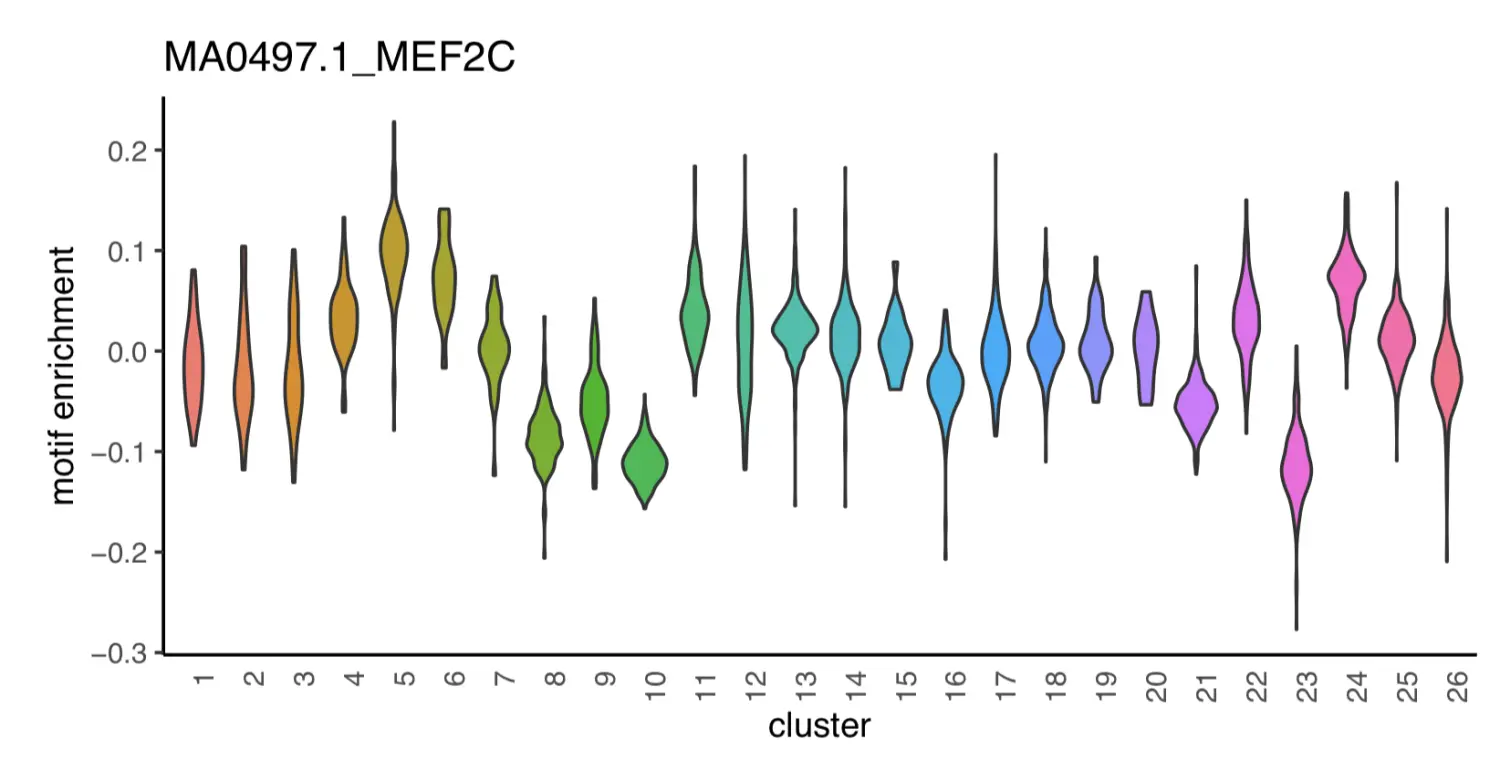
Step 16. GREAT analysis
SnapATAC还可以使用GREAT来识别每个细胞cluster中活跃的生物学通路。在本示例中,我们将首先识别小胶质细胞(cluster 13)中的差异可及性区域DARs,并展示使用GREAT analysis识别出的top 6个GO term的富集情况。
## install R package rGREAT
if (!requireNamespace("BiocManager", quietly=TRUE))
install.packages("BiocManager")
BiocManager::install("rGREAT")
## or install the latest version
library(devtools)
install_github("jokergoo/rGREAT")
library(rGREAT);
DARs = findDAR(
obj=x.sp,
input.mat="pmat",
cluster.pos=13,
cluster.neg.method="knn",
test.method="exactTest",
bcv=0.1, #0.4 for human, 0.1 for mouse
seed.use=10
);
DARs$FDR = p.adjust(DARs$PValue, method="BH");
idy = which(DARs$FDR < 5e-2 & DARs$logFC > 0);
job = submitGreatJob(
gr = x.sp@peak[idy],
bg = NULL,
species = "mm10",
includeCuratedRegDoms = TRUE,
rule = "basalPlusExt",
adv_upstream = 5.0,
adv_downstream = 1.0,
adv_span = 1000.0,
adv_twoDistance = 1000.0,
adv_oneDistance = 1000.0,
request_interval = 300,
max_tries = 10,
version = "default",
base_url = "http://great.stanford.edu/public/cgi-bin"
);
job
## Submit time: 2019-09-04 14:14:02
## Version: default
## Species: mm10
## Inputs: 25120 regions
## Background: wholeGenome
## Model: Basal plus extension
## Proximal: 5 kb upstream, 1 kb downstream,
## plus Distal: up to 1000 kb
## Include curated regulatory domains
##
## Enrichment tables for following ontologies have been downloaded:
## None
运行完所提交的job后,我们可以提取GREAT分析的结果。第一个主要的分析结果是一个富集统计的信息表。默认情况下,它将检索来自三种GO(包括MF,BP,CC)terms的结果和pathway的注释结果。所有的表中都包含所有terms 的统计信息,无论它们是否具有显著性。并且,用户可以通过自定义的cutoff 进行筛选。
tb = getEnrichmentTables(job);
names(tb);
## [1] "GO Molecular Function" "GO Biological Process" "GO Cellular Component"
GBP = tb[["GO Biological Process"]];
head(GBP[order(GBP[,"Binom_Adjp_BH"]),1:5]);
## ID name Binom_Genome_Fraction
## 1 GO:0002376 immune system process 0.12515840
## 2 GO:0002682 regulation of immune system process 0.09012561
## 3 GO:0009987 cellular process 0.80870120
## 4 GO:0048518 positive regulation of biological process 0.43002240
## 5 GO:0050789 regulation of biological process 0.68873070
## 6 GO:0050794 regulation of cellular process 0.66837300
## Binom_Expected Binom_Observed_Region_Hits
## 1 3095.918 5592
## 2 2229.347 4148
## 3 20004.030 22241
## 4 10637.030 13697
## 5 17036.440 19871
## 6 16532.870 19356
参考来源:https://gitee.com/booew/SnapATAC/blob/master/examples/10X_brain_5k/README.md

若有收获,就点个赞吧
0 人点赞
