简介
scRepertoire包是由华盛顿大学的Nick Borcherding博士开发,是一款针对10X V(D)J数据的免疫组库分析工具。它可以直接读取10X cellranger的输出结果,处理该数据并根据两个配对TCR或Ig链分配克隆型,分析colonotype的动态特征,提供了丰富的可视化展示工具。同时,还可以与Seurat、SingleCellExperiment 或 Monocle 3 包的单细胞mRNA 表达数据进行整合分析。
安装并加载所需的R包
BiocManager::install("scRepertoire")# Most up-to-date versiondevtools::install_github("ncborcherding/scRepertoire@dev")# 加载所需的R包suppressMessages(library(scRepertoire))suppressMessages(library(Seurat))# 查看R包的版本,最新的为‘1.3.1’packageVersion("scRepertoire")#[1] ‘1.3.1’
1. 加载并预处理VDJ重叠群数据
我们可以使用scRepertoire包直接读取10x cellranger vdj程序生成的filtered_contig_annotations.csv文件,为每个样本加载该文件并构建一个重叠群(contig)的列表。
# 读取不同的样本contig文件
S1 <- read.csv(".../Sample1/outs/filtered_contig_annotations.csv")
S2 <- read.csv(".../Sample2/outs/filtered_contig_annotations.csv")
S3 <- read.csv(".../Sample3/outs/filtered_contig_annotations.csv")
S4 <- read.csv(".../Sample4/outs/filtered_contig_annotations.csv")
# 构建重叠群列表
contig_list <- list(S1, S2, S3, S4)
这里我们使用scRepertoire包自带的一个数据集进行演示,该数据集来源于三名肾透明细胞癌患者的T细胞,样本由成对的外周血和肿瘤浸润细胞组成。
# 加载内置数据集
data("contig_list") #the data built into scRepertoire
# 查看数据集中contig的注释信息
head(contig_list[[1]])
## barcode is_cell contig_id high_confidence length
## 1 AAACCTGAGAGCTGGT TRUE AAACCTGAGAGCTGGT-1_contig_1 TRUE 705
## 2 AAACCTGAGAGCTGGT TRUE AAACCTGAGAGCTGGT-1_contig_2 TRUE 502
## 3 AAACCTGAGCATCATC TRUE AAACCTGAGCATCATC-1_contig_1 TRUE 693
## 4 AAACCTGAGCATCATC TRUE AAACCTGAGCATCATC-1_contig_2 TRUE 567
## 5 AAACCTGAGCATCATC TRUE AAACCTGAGCATCATC-1_contig_5 TRUE 361
## 6 AAACCTGAGTGGTCCC TRUE AAACCTGAGTGGTCCC-1_contig_1 TRUE 593
## chain v_gene d_gene j_gene c_gene full_length productive cdr3
## 1 TRB TRBV20-1 TRBD1 TRBJ1-5 TRBC1 TRUE TRUE CSASMGPVVSNQPQHF
## 2 TRB None None TRBJ1-5 TRBC1 FALSE None None
## 3 TRB TRBV5-1 TRBD2 TRBJ2-2 TRBC2 TRUE TRUE CASSWSGAGDGELFF
## 4 TRA TRAV12-1 None TRAJ37 TRAC TRUE TRUE CVVNDEGSSNTGKLIF
## 5 TRB None None TRBJ1-5 TRBC1 FALSE None None
## 6 TRB TRBV7-9 TRBD1 TRBJ2-5 TRBC2 TRUE TRUE CASSPSEGGRQETQYF
## cdr3_nt reads umis raw_clonotype_id
## 1 TGCAGTGCTAGCATGGGACCGGTAGTGAGCAATCAGCCCCAGCATTTT 16718 6 clonotype96
## 2 None 6706 3 clonotype96
## 3 TGCGCCAGCAGCTGGTCAGGAGCGGGAGACGGGGAGCTGTTTTTT 26719 11 clonotype97
## 4 TGTGTGGTGAACGATGAAGGCTCTAGCAACACAGGCAAACTAATCTTT 18297 6 clonotype97
## 5 None 882 4 clonotype97
## 6 TGTGCCAGCAGCCCCTCCGAAGGGGGGAGACAAGAGACCCAGTACTTC 11218 6 clonotype98
## raw_consensus_id
## 1 clonotype96_consensus_1
## 2 None
## 3 clonotype97_consensus_2
## 4 clonotype97_consensus_1
## 5 None
## 6 clonotype98_consensus_1
注意:有些VDJ数据处理程序会在生成的细胞barcode前添加样本的注释信息,如“Sample1_AAACCTGAGAGCTGGT”,此时我们可以使用stripBarcode()函数去除barcode前的注释信息,在使用该函数时有以下参数:
- column:指示barcode所在的列
- connector:指示barcode与前缀注释信息之间的连接符
- num_connects:指示barcode前缀连接的级别,其中X_X_AAACGGGAGATGGCGT-1 == 3, X_AAACGGGAGATGGCGT-1 = 2。
# 去除barcode前缀注释信息
for (i in seq_along(contig_list)) {
contig_list[[i]] <- stripBarcode(contig_list[[i]],
column = 1, connector = "_", num_connects = 3)
}
2. 合并VDJ重叠群,构建TCR克隆型
由于CellRanger默认是输出对TCRA和TCRB链的量化,没有将细胞的barcode与clonotype直接对应起来,因此我们需要使用combineTCR()函数将它们合并到一起,同时我们还可以添加样本和ID信息以防止细胞条形码重复。
combined <- combineTCR(contig_list,
samples = c("PY", "PY", "PX", "PX", "PZ","PZ"),
ID = c("P", "T", "P", "T", "P", "T"),
cells ="T-AB")
combineTCR()函数的输出结果是一个合并重叠群的数据列表,它将每个细胞配对的两条链放在一行,同时还将核苷酸序列 (CTnt)、氨基酸序列 (CTaa)、VDJC 基因序列 (CTgene) 或核苷酸和基因序列的组合 (CTstrict) 合并到一起组成克隆型clonotype进行调用。类似的,对于B细胞,我们可以使用combineBCR()函数进行clonotyep重叠群的合并。
这里,有两个需要注意的地方:
- 每个细胞barcode最多只能有2个序列,如果存在更多的序列,则选择具有最高读数的2个。
- TCR克隆型的严格定义 (CTstrict) 是基于V基因和核苷酸序列>85%的归一化Levenshtein距离。
3. 其他预处理功能
3.1 添加附加变量信息
除了添加sample和ID信息外,我们还可以使用addVariable()函数来添加其他的变量信息。我们只需要设置添加的变量的名称和特定的字符或数字值(变量)。例如,这里我们添加处理和排序样本的批次。
example <- addVariable(combined, name = "batch",
variables = c("b1", "b1", "b2", "b2", "b2", "b2"))
example[[1]][1:5,ncol(example[[1]])] # This is showing the first 5 values of the new column added
## [1] "b1" "b1" "b1" "b1" "b1"
3.2 提取重叠群的子集
同样的,我们还可以使用subsetContig()函数在combineTCR()之后删除特定的列表元素。为了提取相应的重叠群子集信息,我们需要确定要用于子集化的向量(name)以及子集的变量值(variables)。下面我们将从PX和PY样本中分离出4个测序结果。
subset <- subsetContig(combined, name = "sample", variables = c("PX", "PY"))
4. 可视化重叠群
cloneCall参数:
- “gene”:使用包含TCR/Ig的VDJC基因
- “nt”:使用CDR3区域的核苷酸序列
- “aa”:使用CDR3区域的氨基酸序列、
- “gene+nt”:使用包含TCR/Ig的VDJC基因+CDR3区域的核苷酸序列
需要注意的是,TCR克隆型的定义基本上是使用两个基因座的基因或nt/aa序列以及CDR3序列的组合来命名的。在scRepertoire中,克隆型的调用没有考虑CDR3序列中的微小变异。因此,”gene”方法将是最敏感的,而使用”nt”或”aa”方法是中度敏感,”gene+nt”方法对克隆型最具特异性。
探索克隆型的第一个函数是quantContig(),它将返回独特克隆型的总数或相对数量。
scale参数:
- TRUE - 独特克隆型的相对百分比按克隆型库的总大小缩放
- FALSE - 报告独特克隆型的总数
quantContig(combined, cloneCall="gene+nt", scale = TRUE)
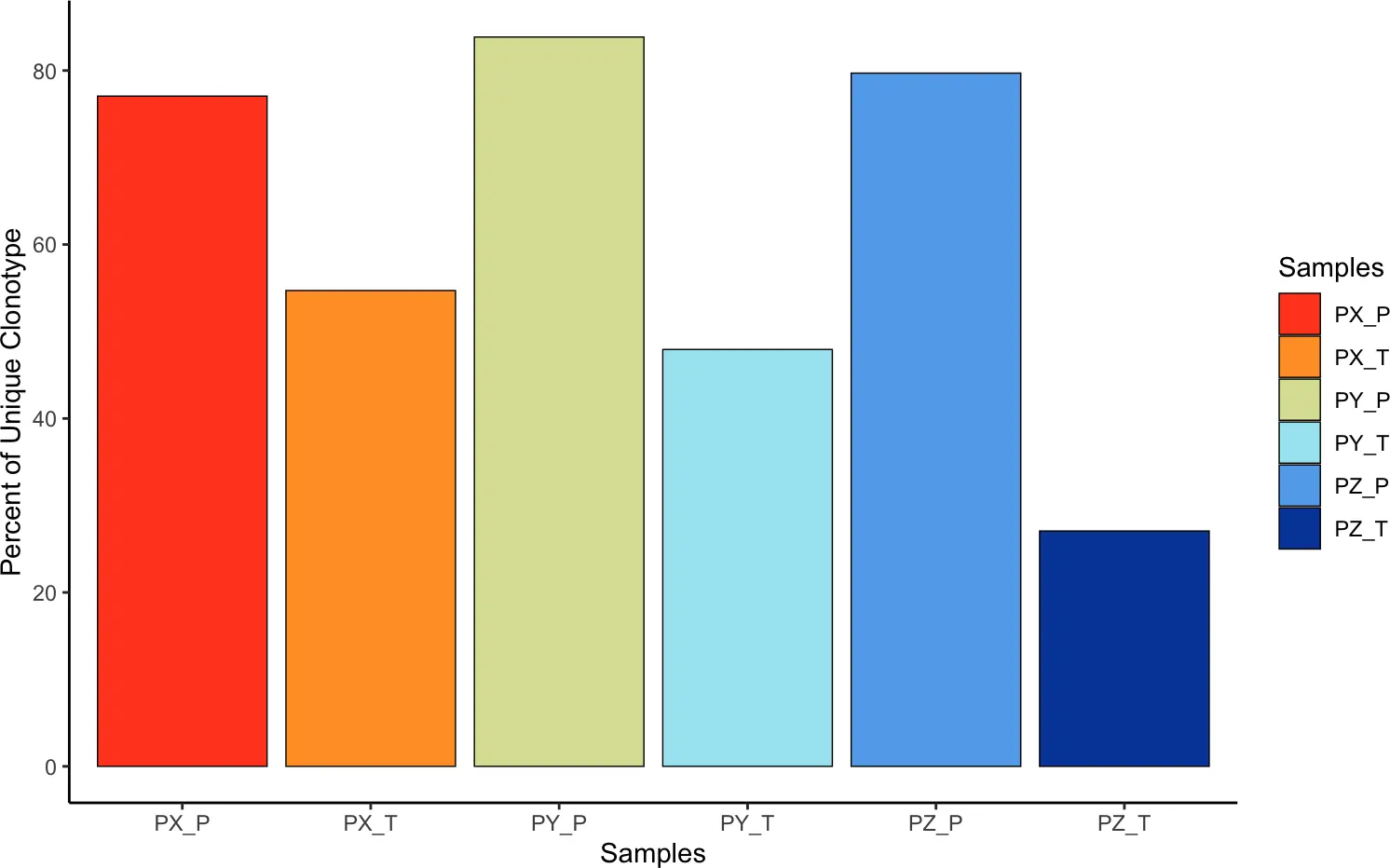
#设置exportTable = T,则输出表格而非图形
quantContig_output <- quantContig(combined, cloneCall="gene+nt",
scale = TRUE, exportTable = TRUE)
quantContig_output
## contigs values total scaled
## 1 2690 PY_P 3208 83.85287
## 2 1495 PY_T 3119 47.93203
## 3 823 PX_P 1068 77.05993
## 4 918 PX_T 1678 54.70799
## 5 1143 PZ_P 1434 79.70711
## 6 749 PZ_T 2768 27.05925
#设置group = "ID",进行分组可视化
quantContig(combined, cloneCall="gene", group = "ID", scale = TRUE)
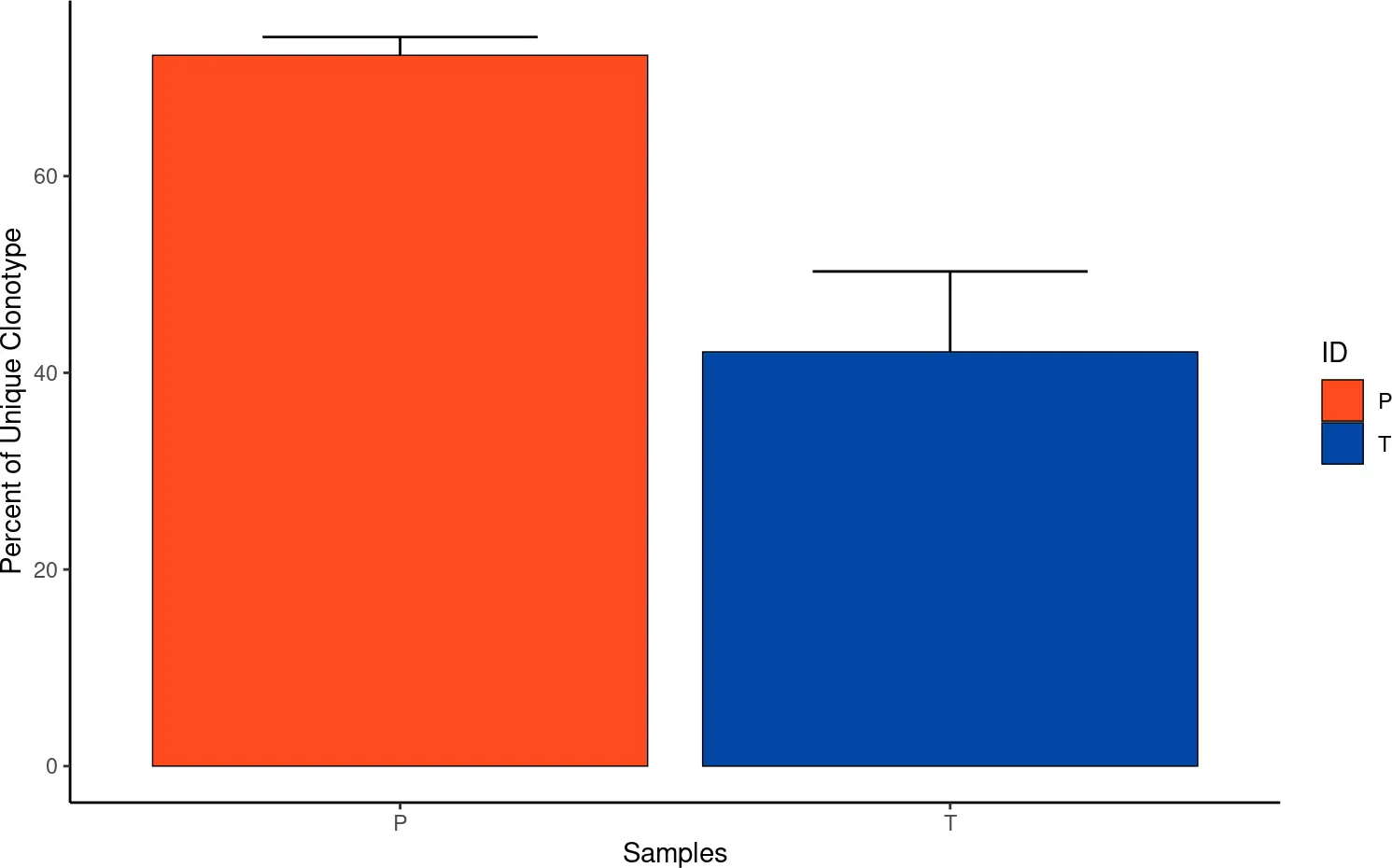
我们还可以使用abundanceContig()函数通过丰度来查看克隆型的相对分布。它将生成一个折线图,展示不同样本中克隆型的总数量。
abundanceContig(combined, cloneCall = "gene", scale = FALSE)
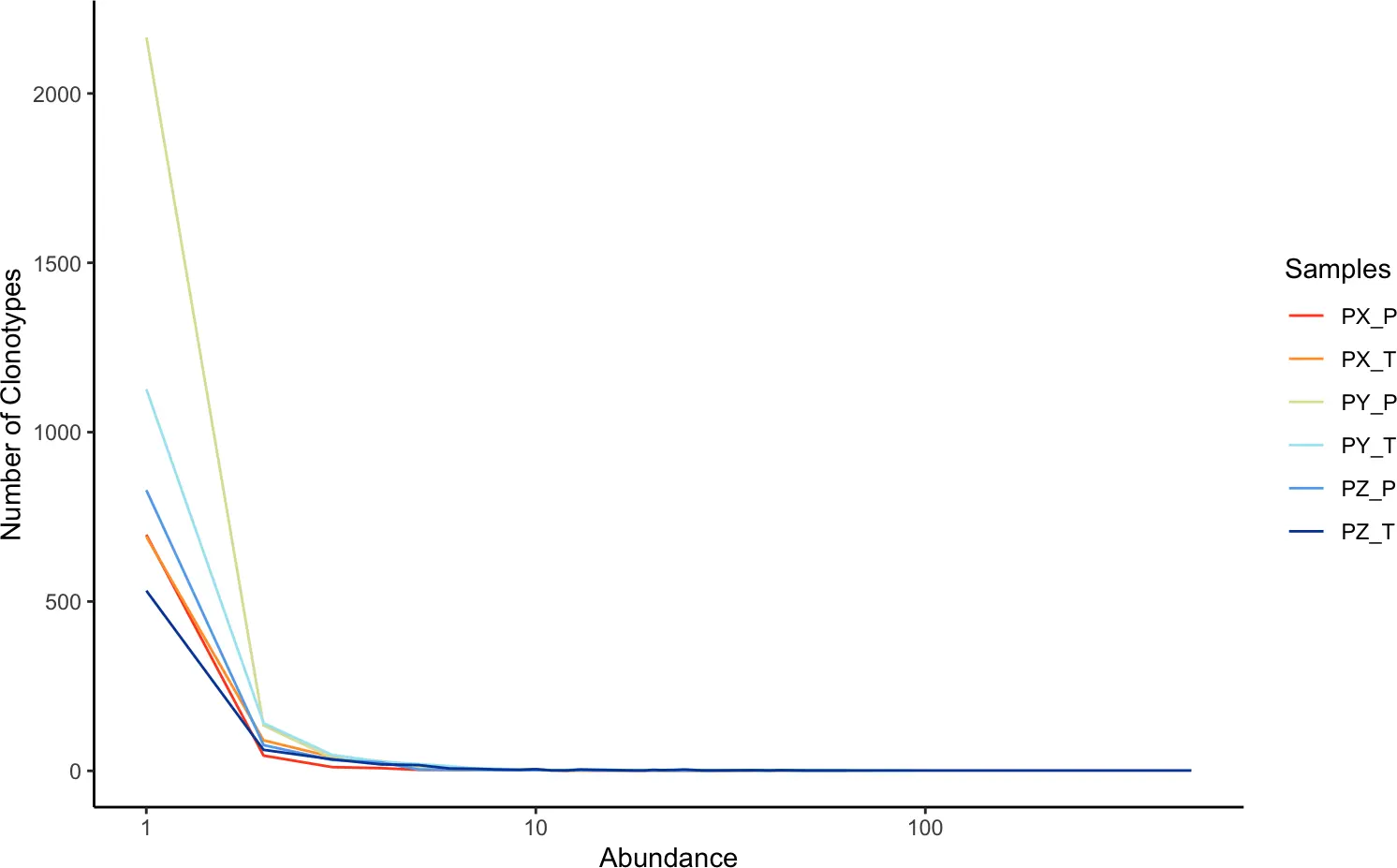
与上面一样,我们也可以使用函数中的group变量根据contig对象中的向量对其进行分组。
abundanceContig(combined, cloneCall = "gene", group = "ID", scale = FALSE)
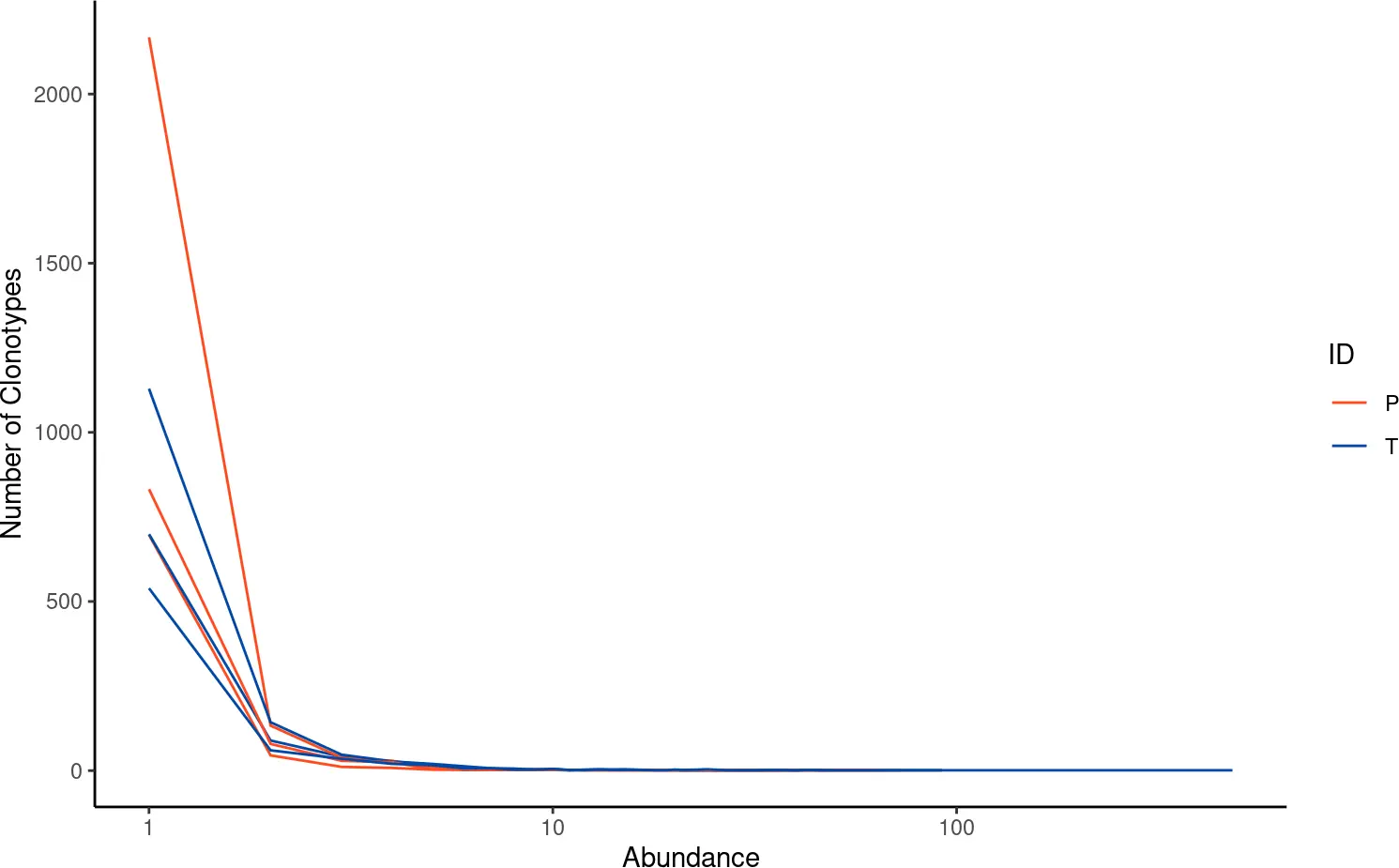
设置exportTable = T,输出相应的表格
abundanceContig(combined, cloneCall = "gene", exportTable = T)
## # A tibble: 7,230 x 3
## CTgene Abundance values
## <chr> <int> <chr>
## 1 NA_TRBV10-1.TRBJ2-2.TRBD1.TRBC2 1 PY_P
## 2 NA_TRBV10-2.TRBJ1-1.TRBD1.TRBC1 1 PY_P
## 3 NA_TRBV10-2.TRBJ2-1.TRBD2.TRBC2 1 PY_P
## 4 NA_TRBV10-2.TRBJ2-3.TRBD2.TRBC2 1 PY_P
## 5 NA_TRBV10-3.TRBJ1-1.None.TRBC1 2 PY_P
## 6 NA_TRBV10-3.TRBJ1-1.TRBD1.TRBC1 2 PY_P
## 7 NA_TRBV10-3.TRBJ1-5.None.TRBC1 1 PY_P
## 8 NA_TRBV10-3.TRBJ1-5.TRBD2.TRBC1 1 PY_P
## 9 NA_TRBV10-3.TRBJ2-1.TRBD1.TRBC2 2 PY_P
## 10 NA_TRBV10-3.TRBJ2-2.TRBD2.TRBC2 1 PY_P
## # … with 7,220 more rows
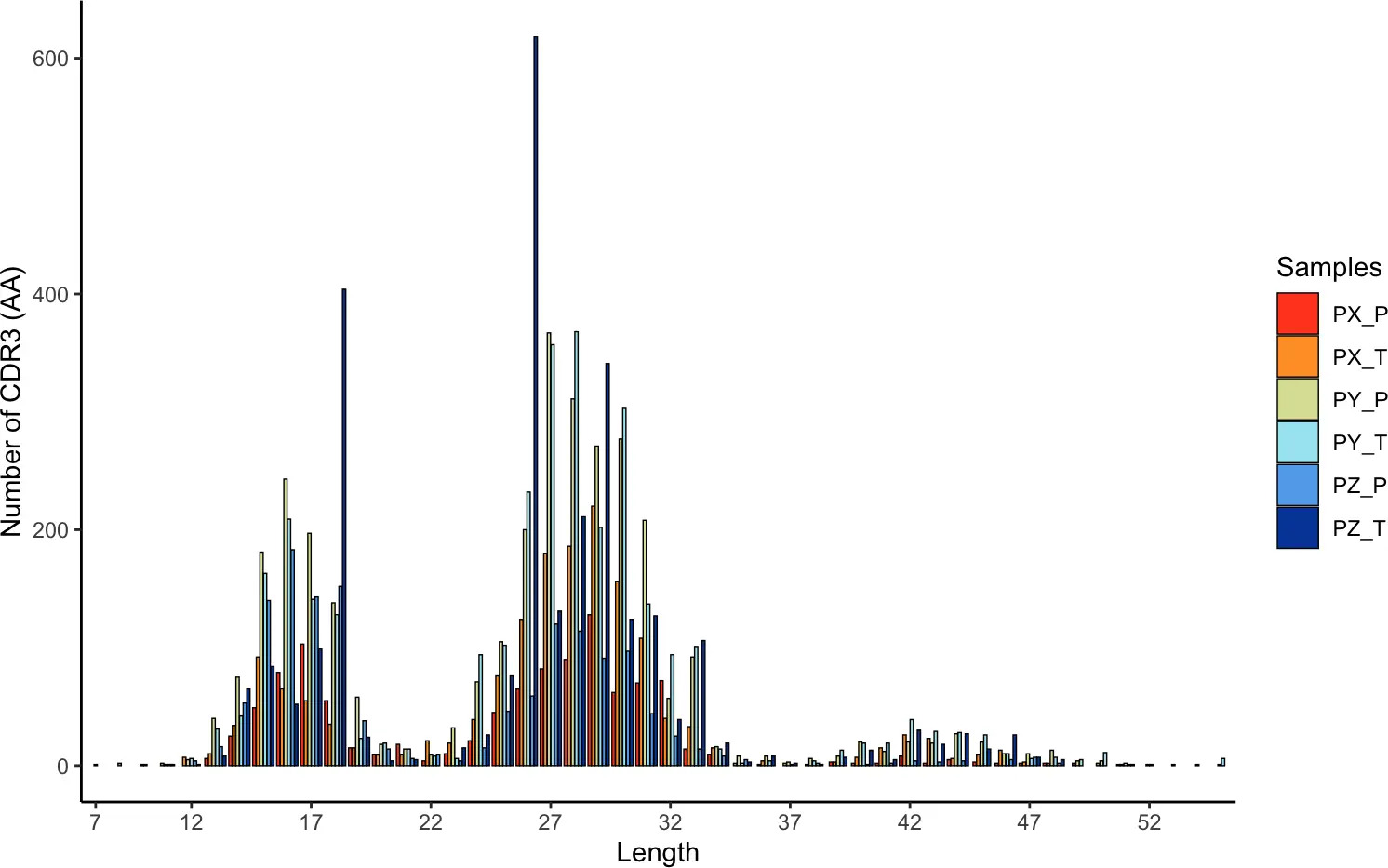
我们可以通过调用lengtheContig()函数来查看CDR3序列的长度分布。重要的是,与其他基本可视化不同,这里cloneCall只能选择“nt”或“aa”。
chain参数:
- “both” 选择组合链进行可视化
- “TRA”、“TRB”、“TRD”、“TRG”、“IGH”或“IGL” 选择特定单链进行可视化
lengthContig(combined, cloneCall="aa", chains = "combined")
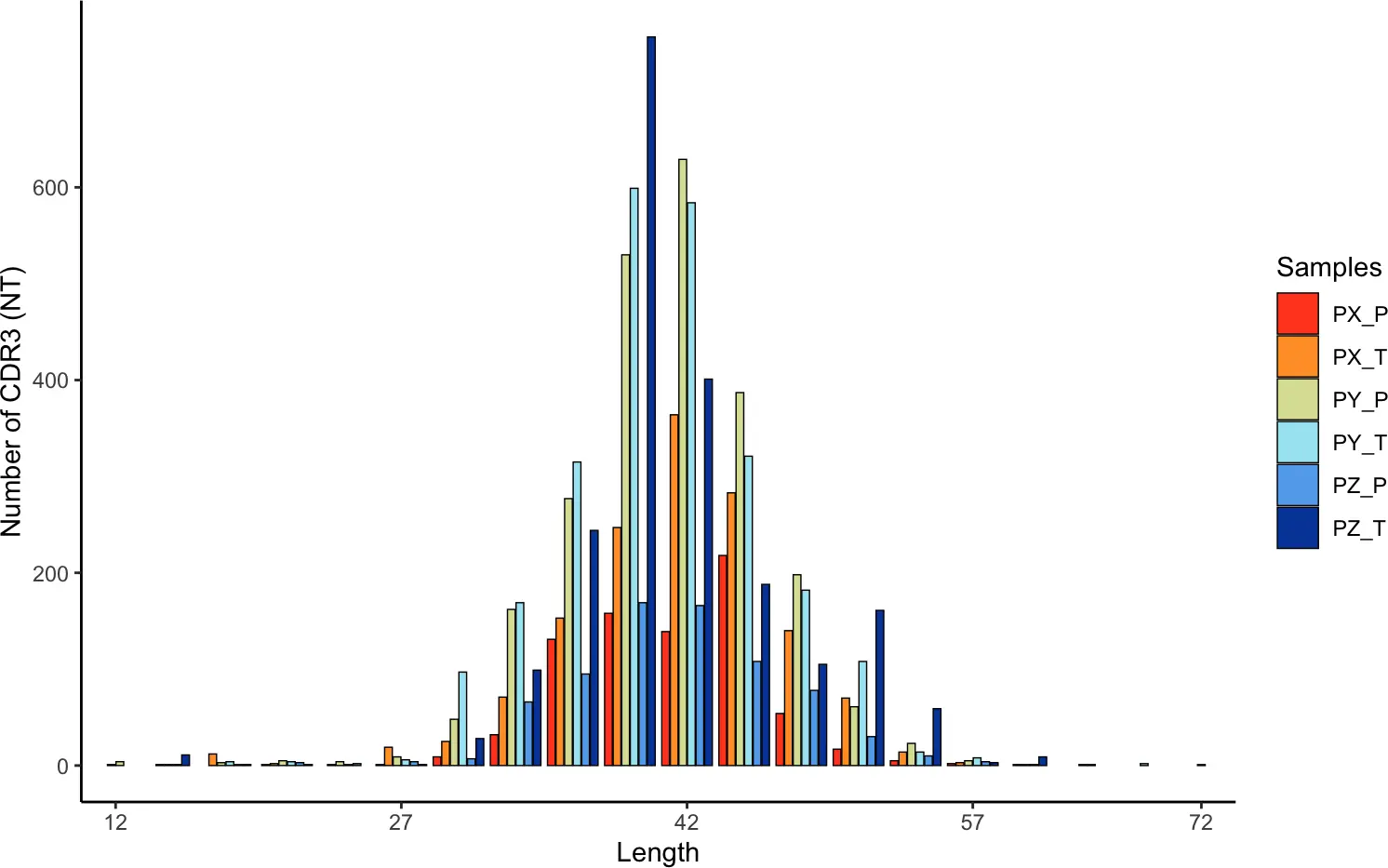
lengthContig(combined, cloneCall="nt", chains = "TRA")
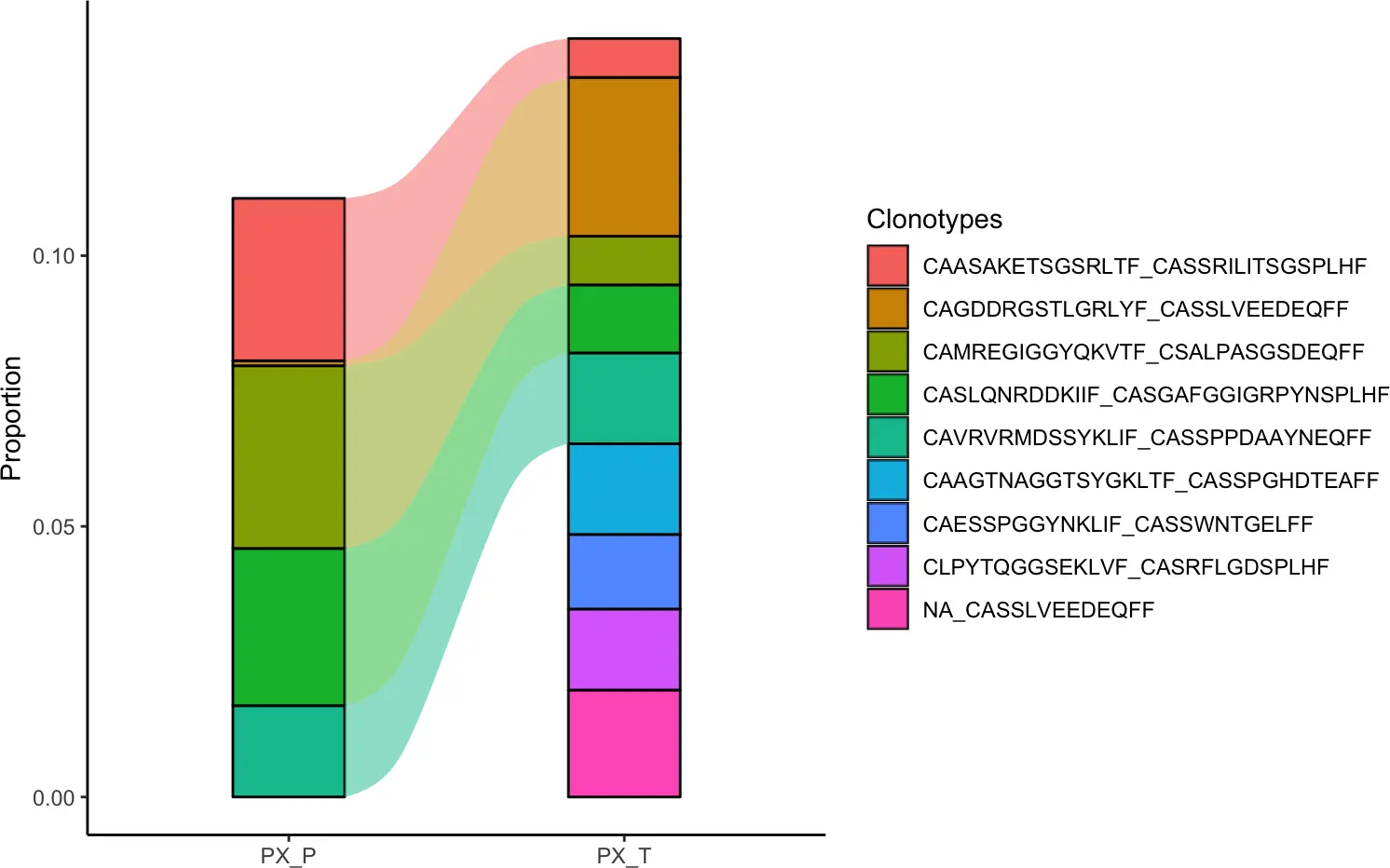
我们还可以使用compareClonotypes()函数查看样本之间的克隆型和动态变化。
可以设置以下参数:
samples
- 设置特定样本的名称
graph
- “alluvial” - 绘制冲击,如下图所
- “area” - 绘制相应克隆型的面积图
number
- 设置要绘的最高克隆型数,这将根据单个样本的频率计算,也可以留空。
clonotypes
- 设置可用于分离特定的克隆型序列,确保调用与您想要可视化的序列相匹配。
compareClonotypes(combined, numbers = 10,
samples = c("PX_P", "PX_T"),
cloneCall="aa", graph = "alluvial")
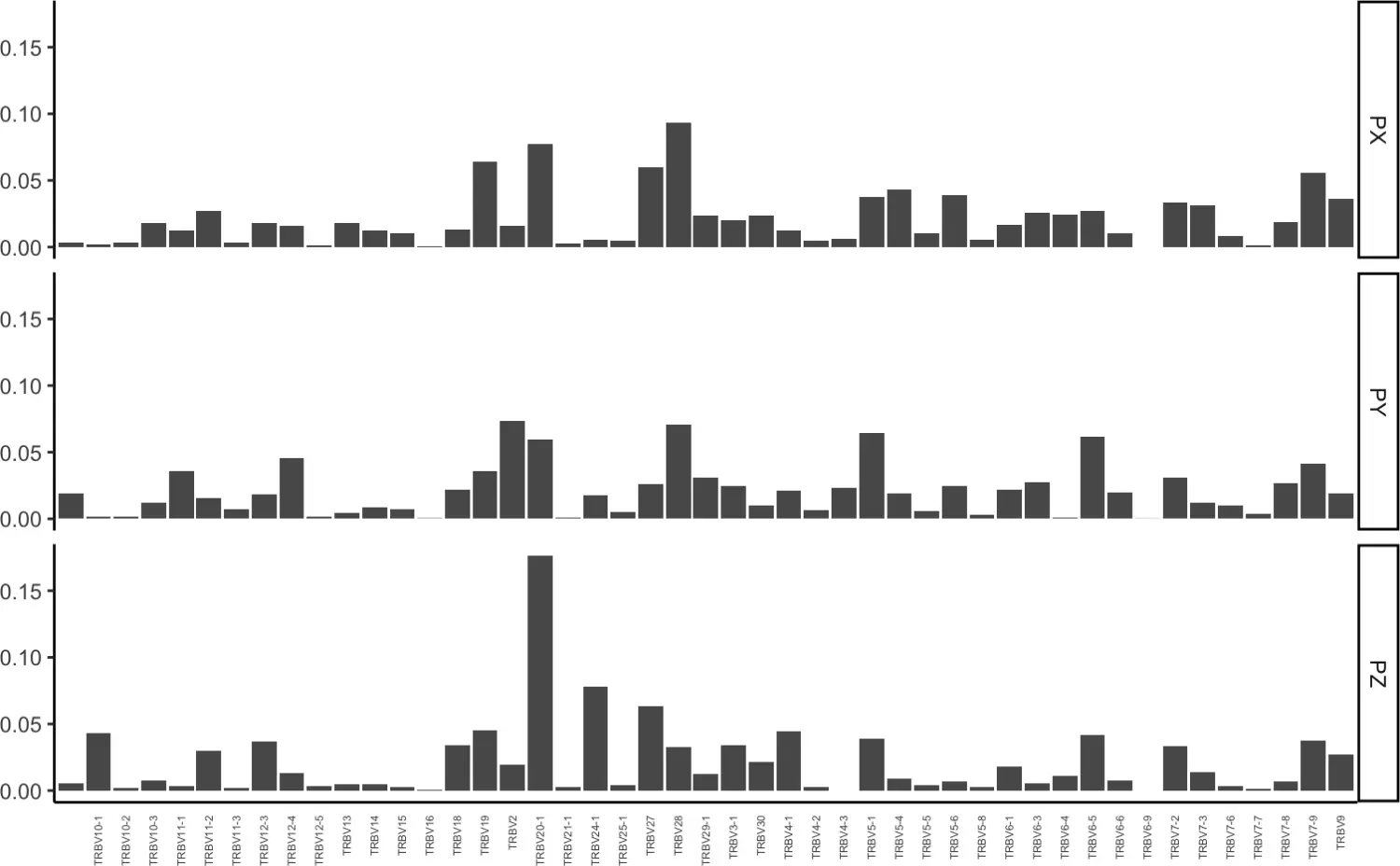
我们还可以使用vizVgenes()函数可视化TCR或BCR基因的相对使用情况。
可以设置以下参数:
gene
- “V”
- “D”
- “J”
- “C”
chain
- “TRB”
- “TRA”
- “TRG”
- “TRD” + “IGH”
- “IGL”
plot
- “bar” for a bar chart
- “heatmap” for a heatmap
separate
Variable to separate the counts along the y-axis
scale
- TRUE to scale the graph by number of genes per sample
- FALSE to report raw numbers
vizVgenes(combined, TCR="TCR1", facet.x = "sample", facet.y = "ID")
5. 更多高级克隆型分析
5.1 Clonal Space Homeostasis克隆空间稳态
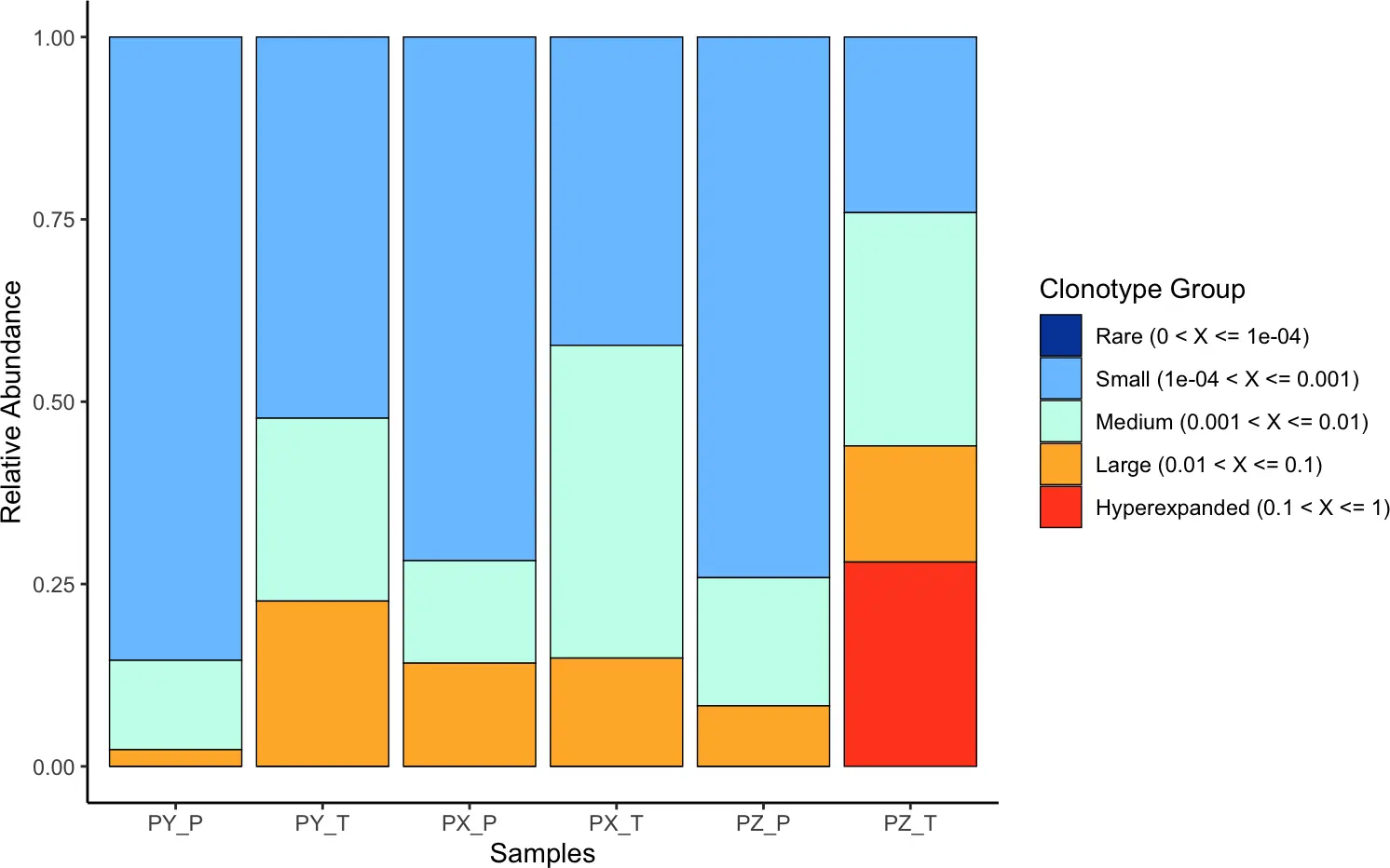
我们可以使用clonalHomeostasis()函数查看不同克隆型在整个克隆空间内的相对丰度,该分析将克隆型分为rare, small, medium, large和hyperexpanded 5大类,并统计各个类别的占比。
cloneTypes:
- Rare = .0001
- Small = .001
- Medium = .01
- Large = .1
- Hyperexpanded = 1
clonalHomeostasis(combined, cloneCall = "gene")
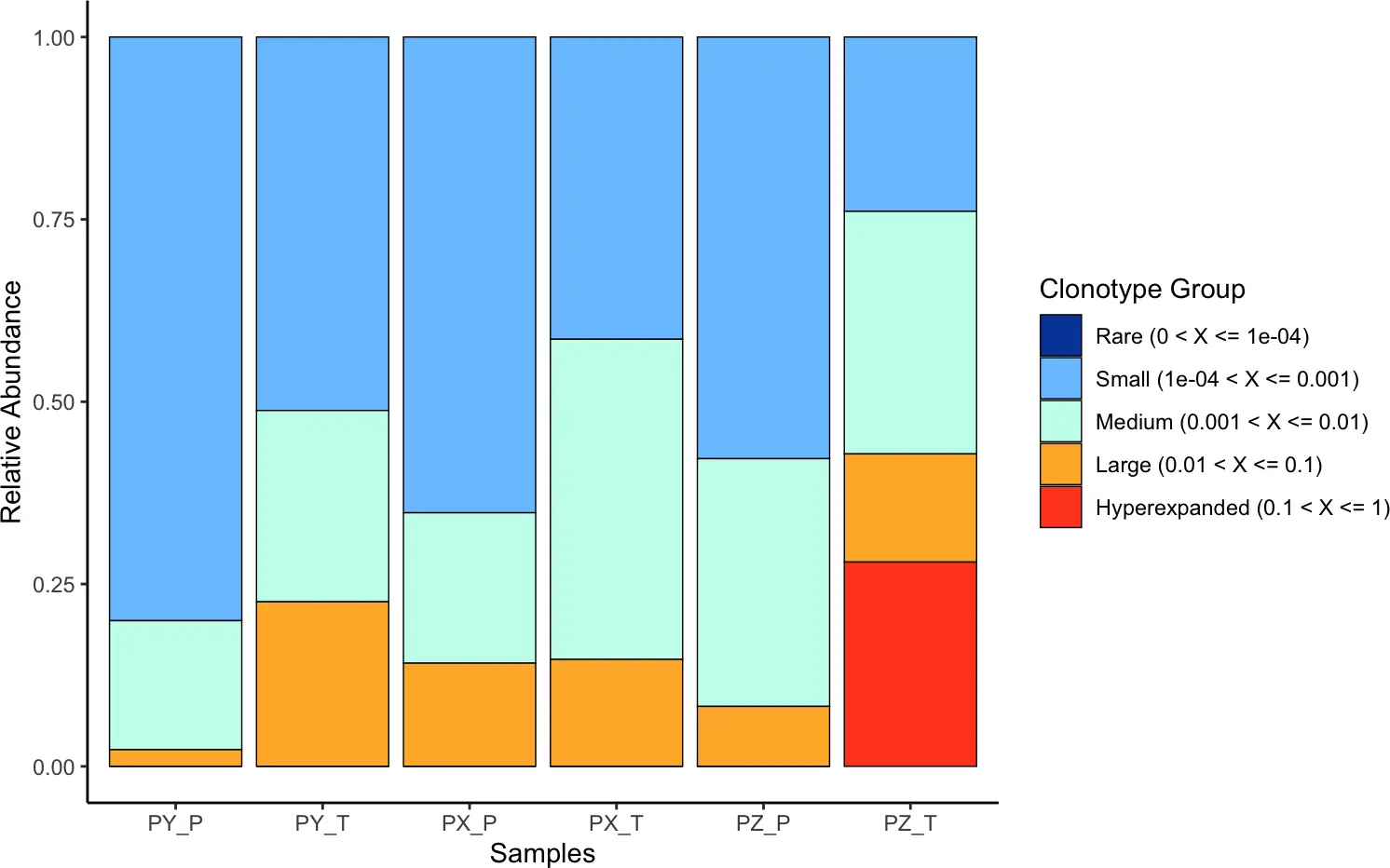
clonalHomeostasis(combined, cloneCall = "aa")
5.2 Clonal Proportion克隆型比例
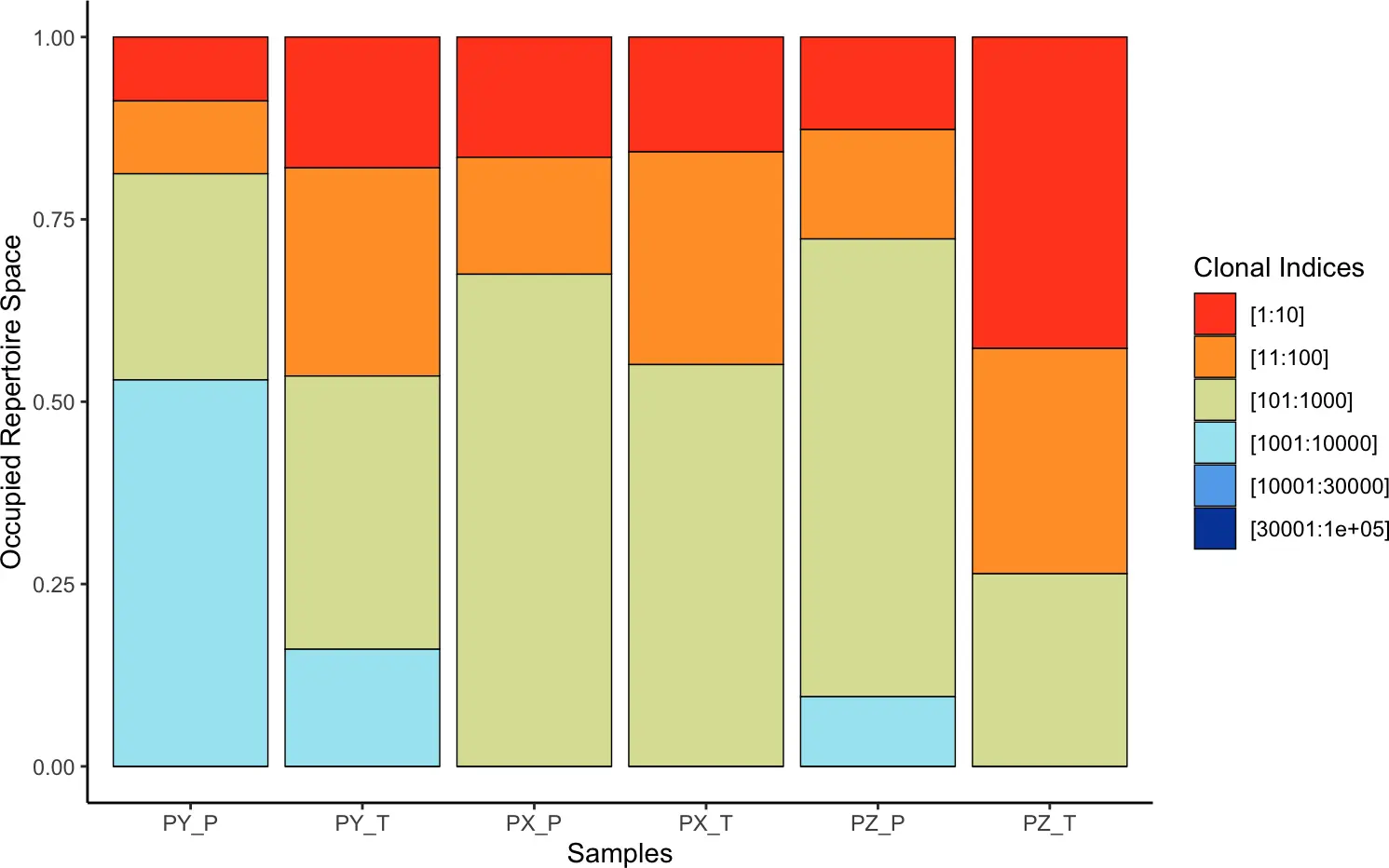
类似的,我们还可以使用clonalProportion()函数查看克隆型的比例,按克隆型的出现频率将其进行排名,1:10表示每个样本中的前10个克隆型。
split:
- 10
- 100
- 1000
- 10000 30000 100000
clonalProportion(combined, cloneCall = "gene")
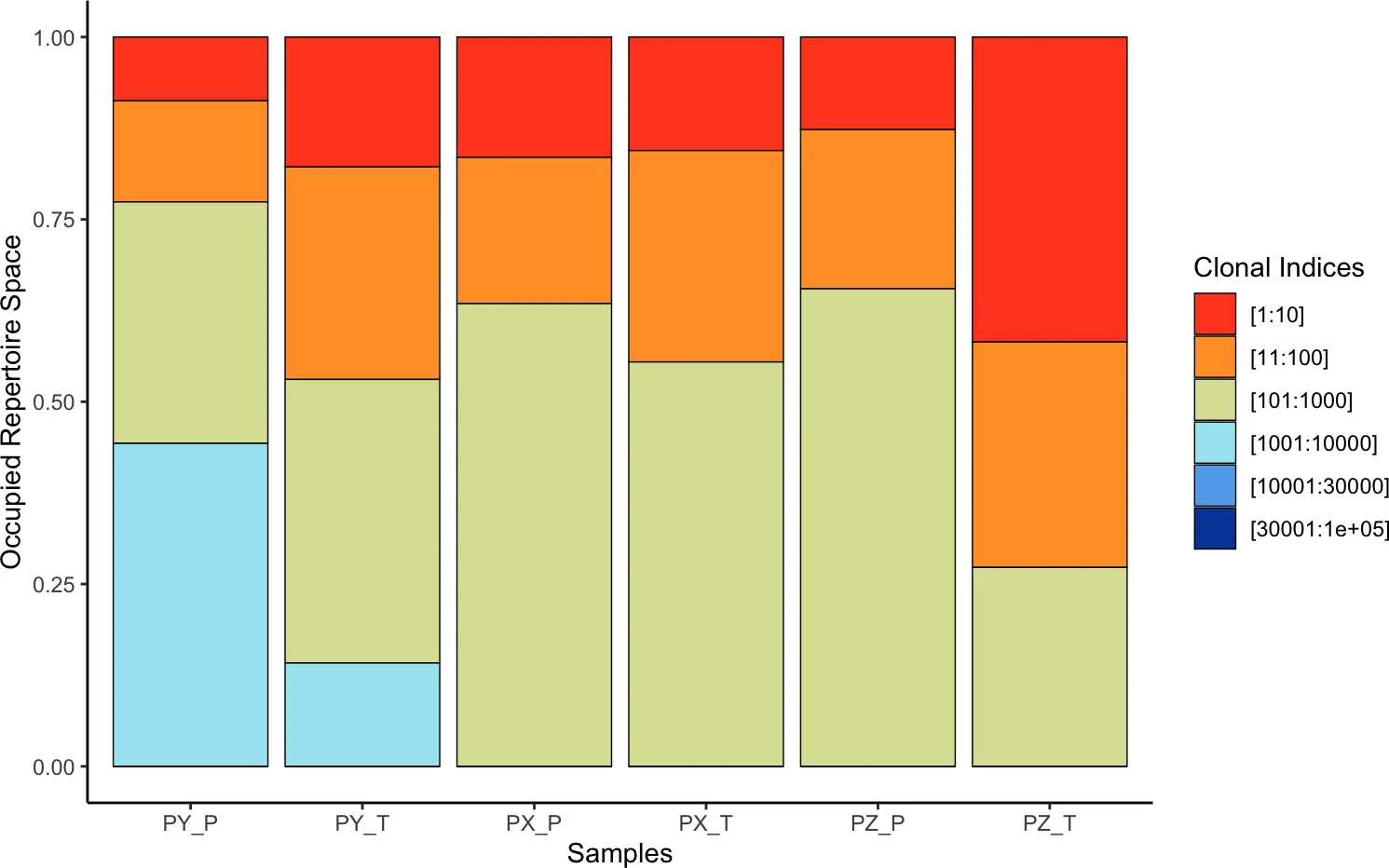
clonalProportion(combined, cloneCall = "nt")
5.3 Overlap Analysis重叠分析
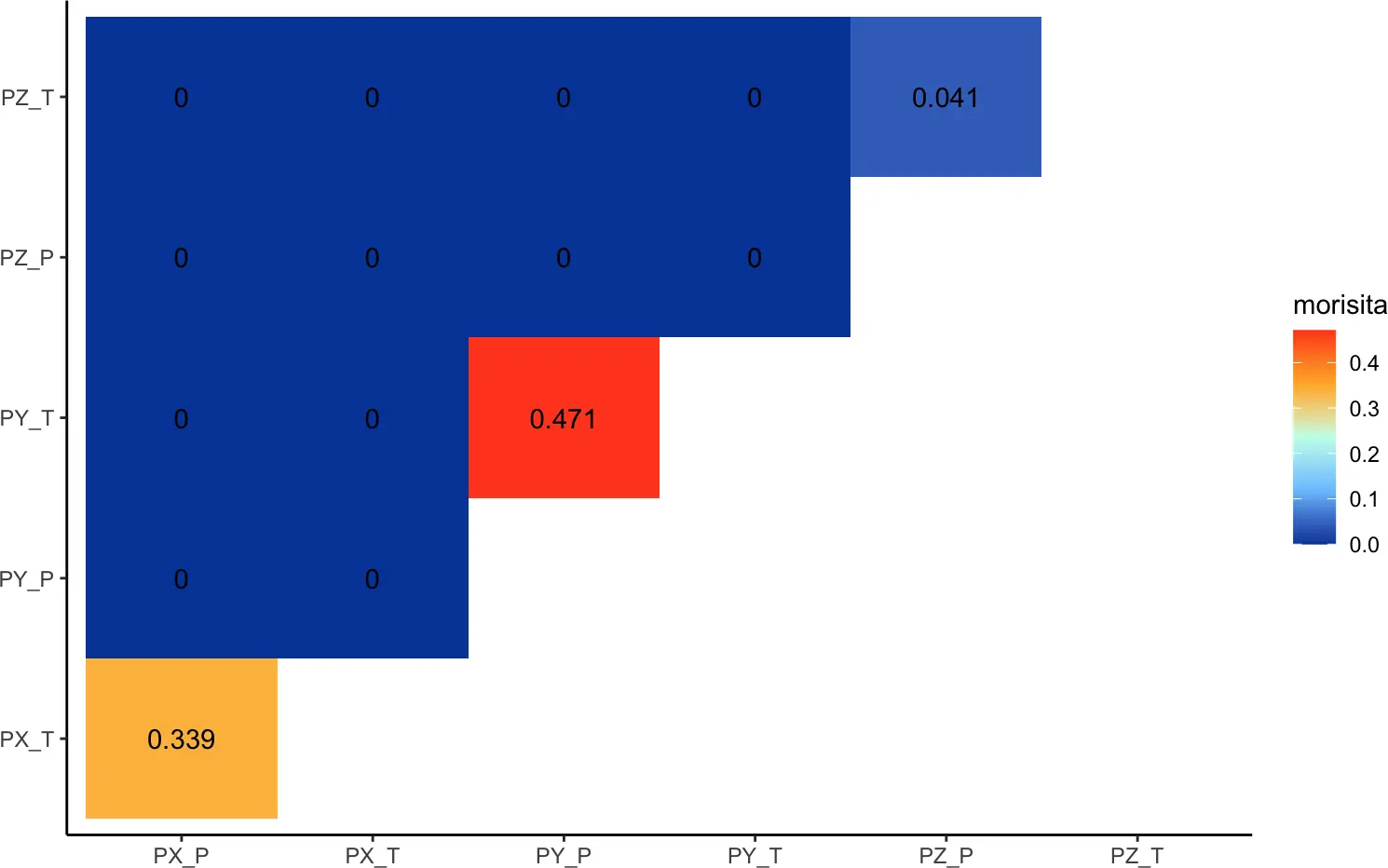
我们可以使用clonalOverlap()函数分析样本之间的相似性,目前有三种方法可用于clonalOverlap():
1)overlap coefficient,2)Morisita index和3)Jaccard index。
clonalOverlap(combined, cloneCall = "gene+nt", method = "morisita")
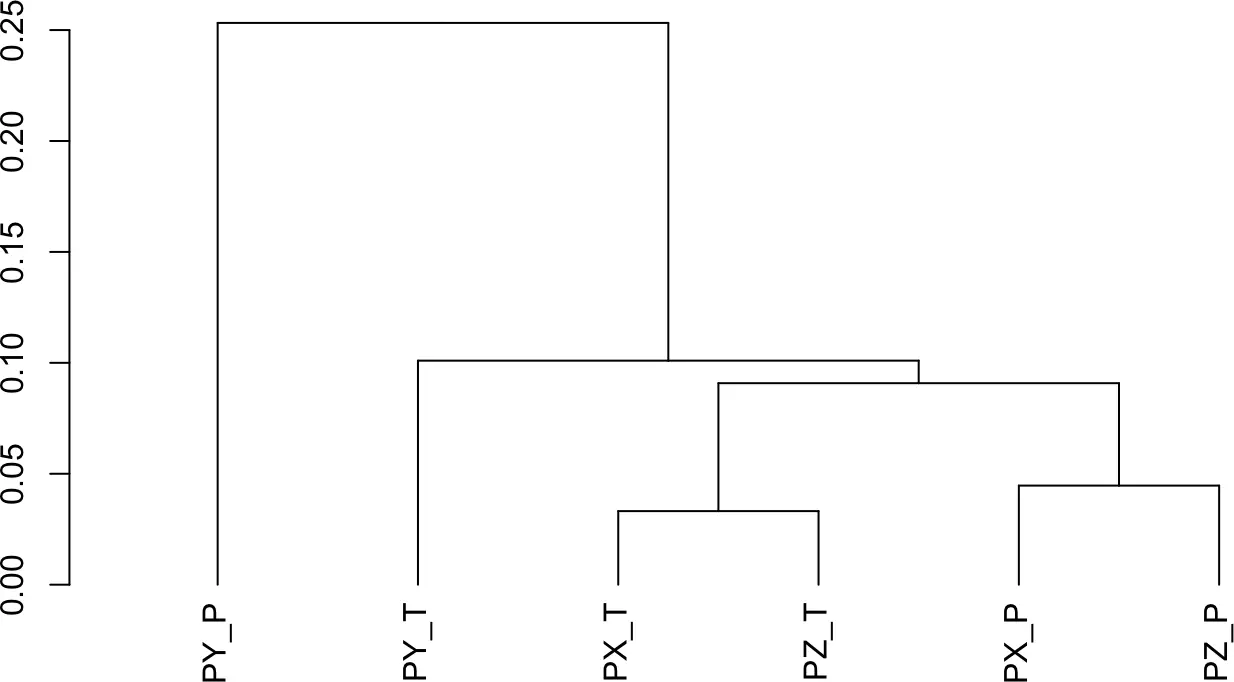
我们还可以使用 clonesizeDistribution()函数通过克隆大小分布对样本进行聚类,该函数改编自powerTCR包。
clonesizeDistribution(combined, cloneCall = "gene+nt",
method="ward.D2")
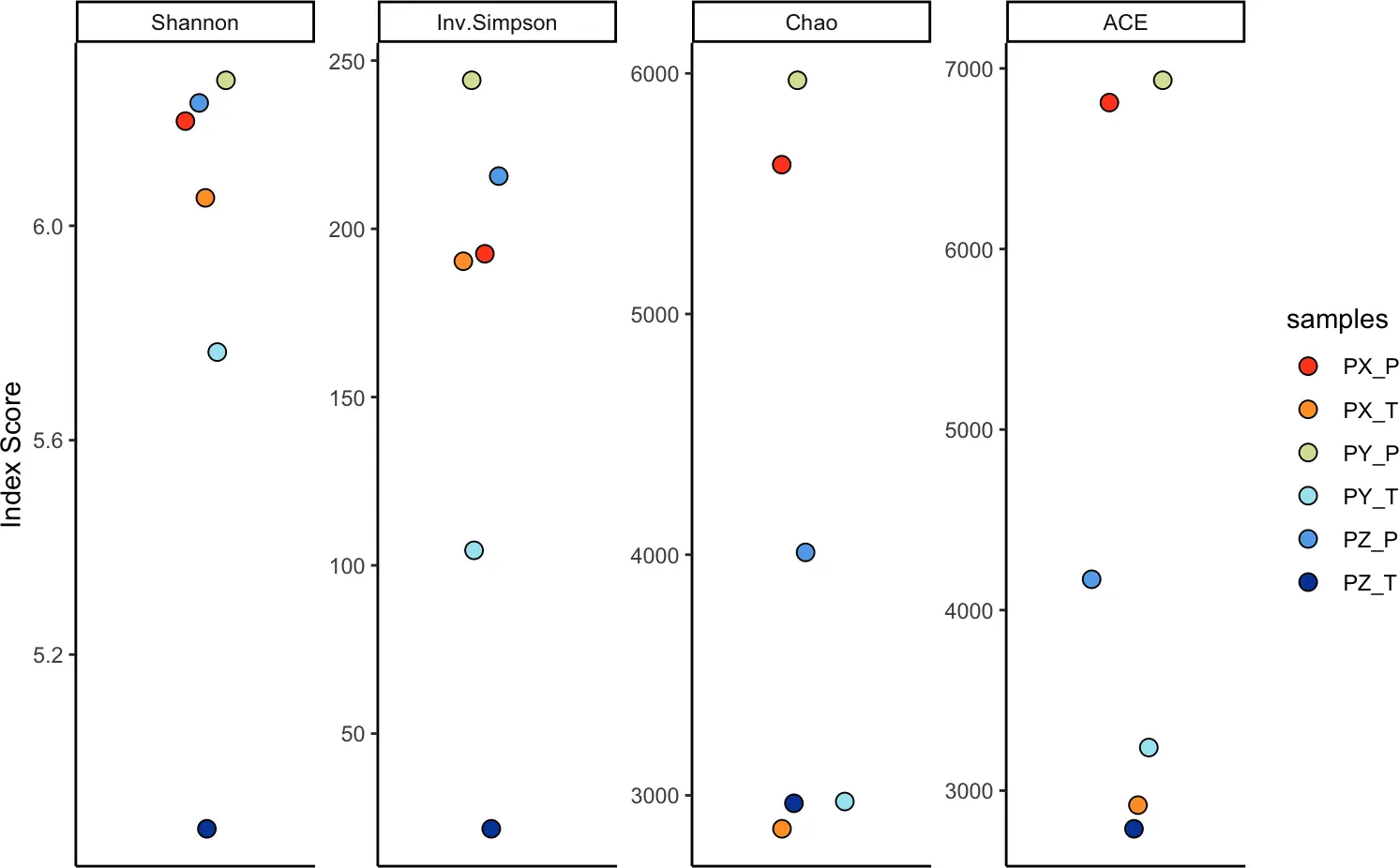
5.4 Diversity Analysis多样性分析
多样性也可以通过样本或其他变量来衡量。多样性可以通过以下四个指标来进行计算:
1)Shannon, 2) inverse Simpson, 3) Chao1和4)based Coverage Estimator (ACE)。
前两者通常用于估计基线多样性,而Chao/ACE指数用于估计样本的丰富度。此函数的新实现包括使用最少数量的独特克隆型对 100 个引导带 (n.boots) 进行下采样,作为更稳健的多样性估计。
clonalDiversity(combined, cloneCall = "gene", group = "samples",
n.boots = 100)
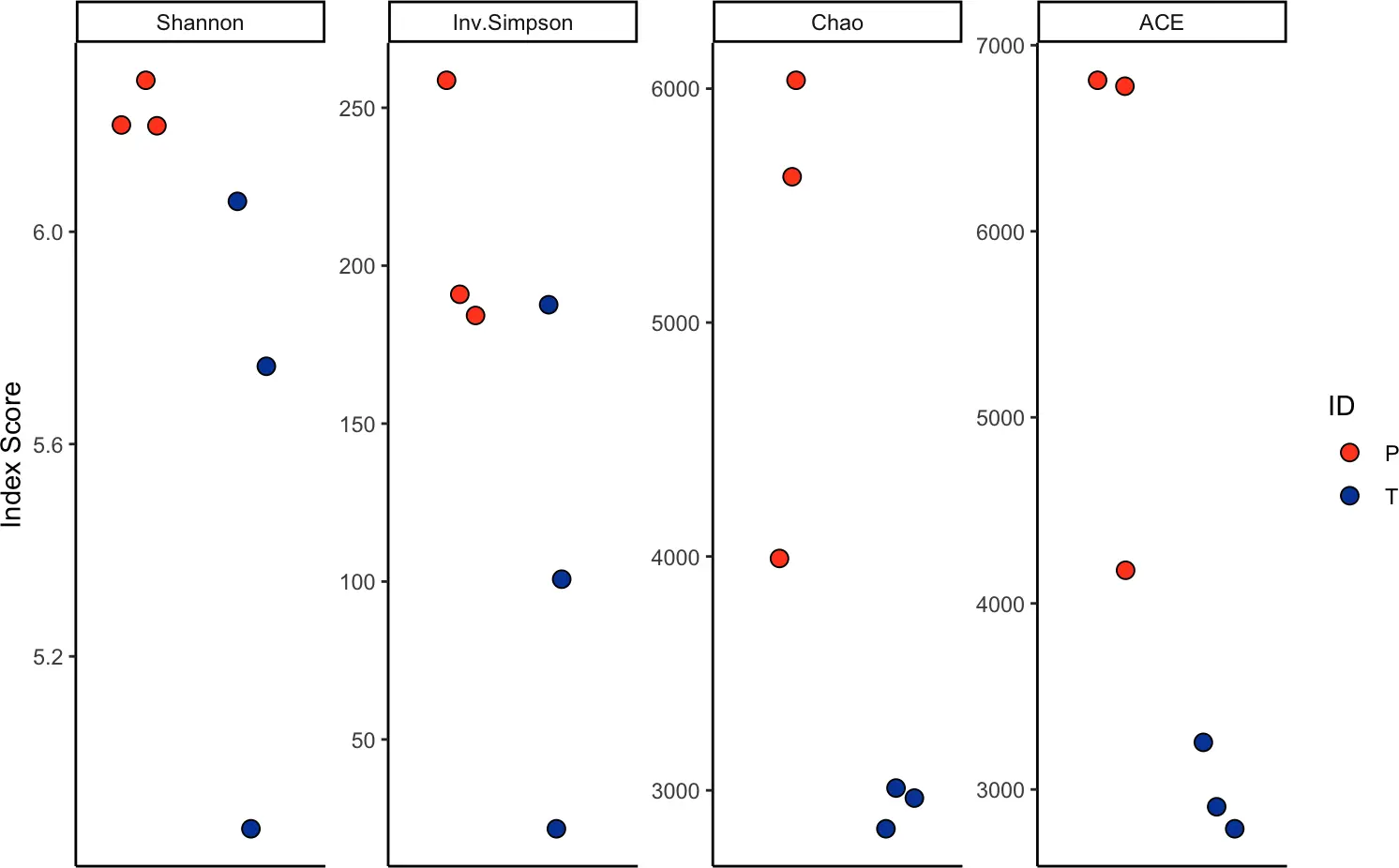
clonalDiversity(combined, cloneCall = "gene", group = "ID")
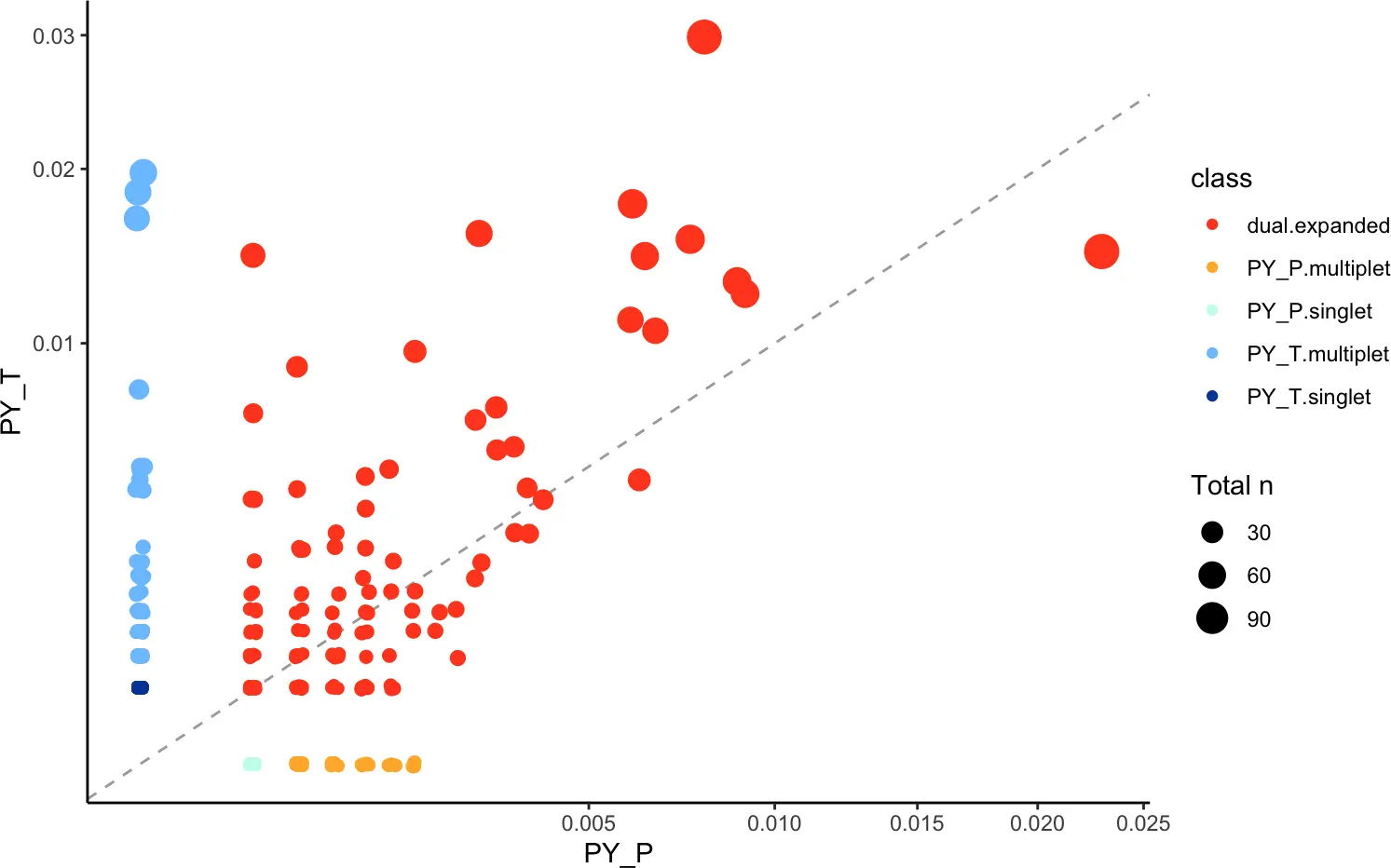
5.5 Scatter Compare散点图比较(仅限开发版)
我们可以使用scatterClonotype()函数计算两个样本的相对克隆型,并生成比较两个样本的散点图。
可以设置以下参数:
x.axis and y.axis:
- 设置 x 轴和 y 轴上的列表元素的名称,例如“PX_P”和“PX_T”。
dot.size:
- “total” 显示 x 轴和 y 轴之间的克隆总数
- Name of the list element to use for size calculation
graph:
- “proportion” 表示克隆型在所有克隆型中代表的相对比例
- “count” 表示按样本的克隆型总数
scatterClonotype(combined, cloneCall ="gene",
x.axis = "PY_P",
y.axis = "PY_T",
dot.size = "total",
graph = "proportion")
5.6 Clustering Clonotypes克隆型聚类
通过检查序列的编辑距离,我们可以使用clusterTCR()函数对链的核苷酸或氨基酸序列进行克隆型的聚类分析。
sub_combined <- clusterTCR(combined[[2]], chain = "TCRA",
sequence = "aa",
threshold = 0.85, group = NULL)
sub_combined <- as.data.frame(sub_combined)
counts_TCRAcluster <- table(sub_combined$TCRA_cluster)
counts_TCRA<- table(sub_combined$cdr3_aa1)
#Change in histogram range using clusters over exact amino acid sequence
plot(rev(sort(counts_TCRAcluster)), xaxt='n')

plot(rev(sort(counts_TCRA)), xaxt='n')

6. 与Seurat包进行整合分析
除了对单细胞免疫组库数据进行常规分析外,我们还可以使用scRepertoire包将其与scRNA-seq数据进行整合分析。通过细胞barcode信息将VDJ数据和scRNA-seq的基因表达数据联系起来,就可以将clonotype显示在降维图上,也可以基于细胞聚类后的cluster展示clonotype的分布情况。
这里,我们利用官网提供的示例scRNA-seq数据演示,它是一个降维聚类后的seurat对象。
# 加载示例scRNA-seq数据
seurat <- get(load("/Users/data/seurat2.rda"))
# 查看细胞降维聚类信息
DimPlot(seurat, label = T) + NoLegend()
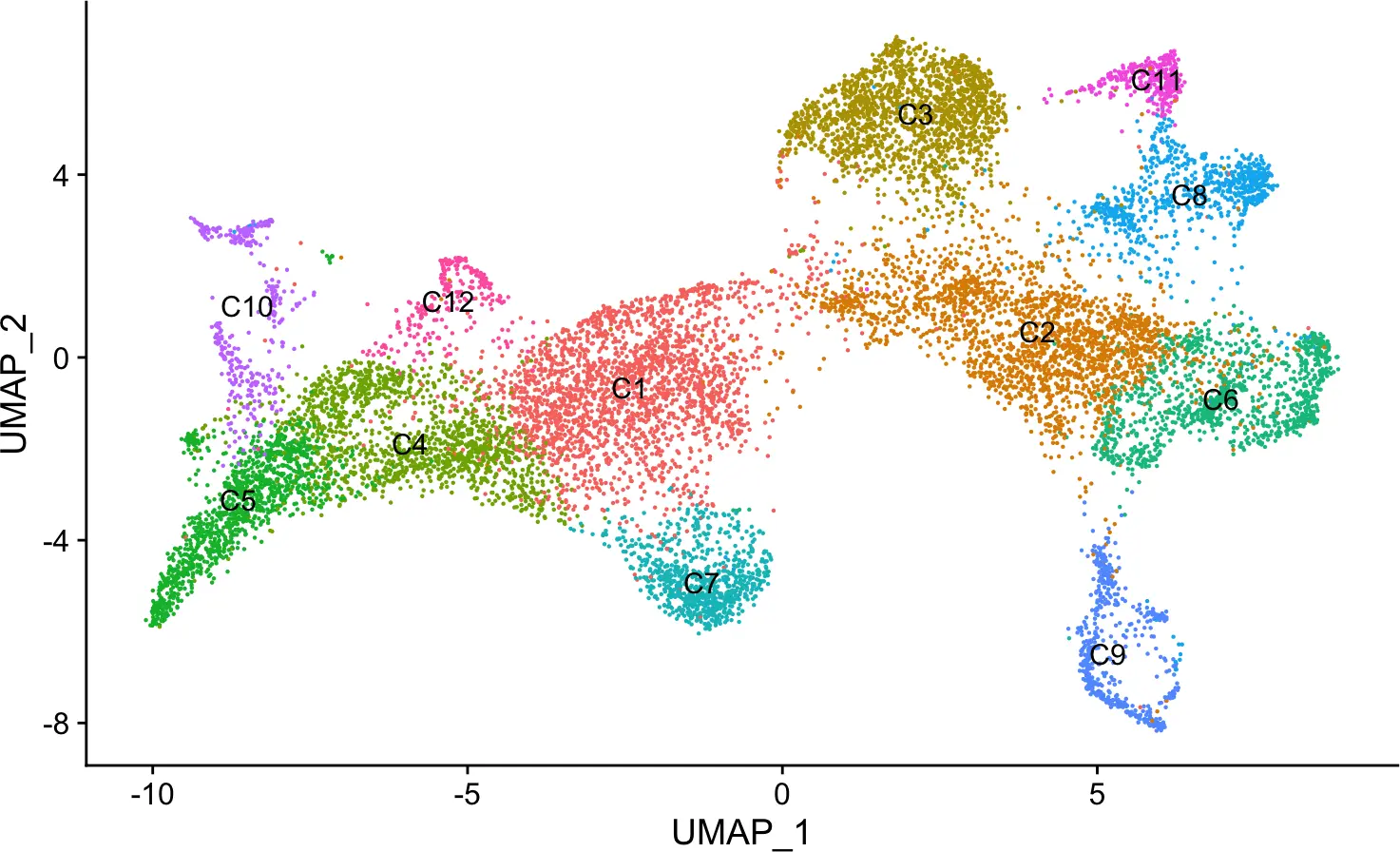
table(Idents(seurat))
## C1 C2 C3 C4 C5 C6 C7 C8 C9 C10 C11 C12
## 2293 2138 1746 1419 1167 1128 807 792 495 357 328 241
接下来,我们可以使用combineExpression()函数获取克隆型信息,并将其附加到我们的Seurat对象中。需要注意的是,该函数要求匹配的colontype的细胞barcode要与Seurat对象中的细胞行名相一致。如果匹配不一致,可能会导致结合失败。
seurat <- combineExpression(combined, seurat,
cloneCall="gene",
groupBy = "sample",
proportion = FALSE,
cloneTypes=c(Single=1, Small=5, Medium=20, Large=100, Hyperexpanded=500))
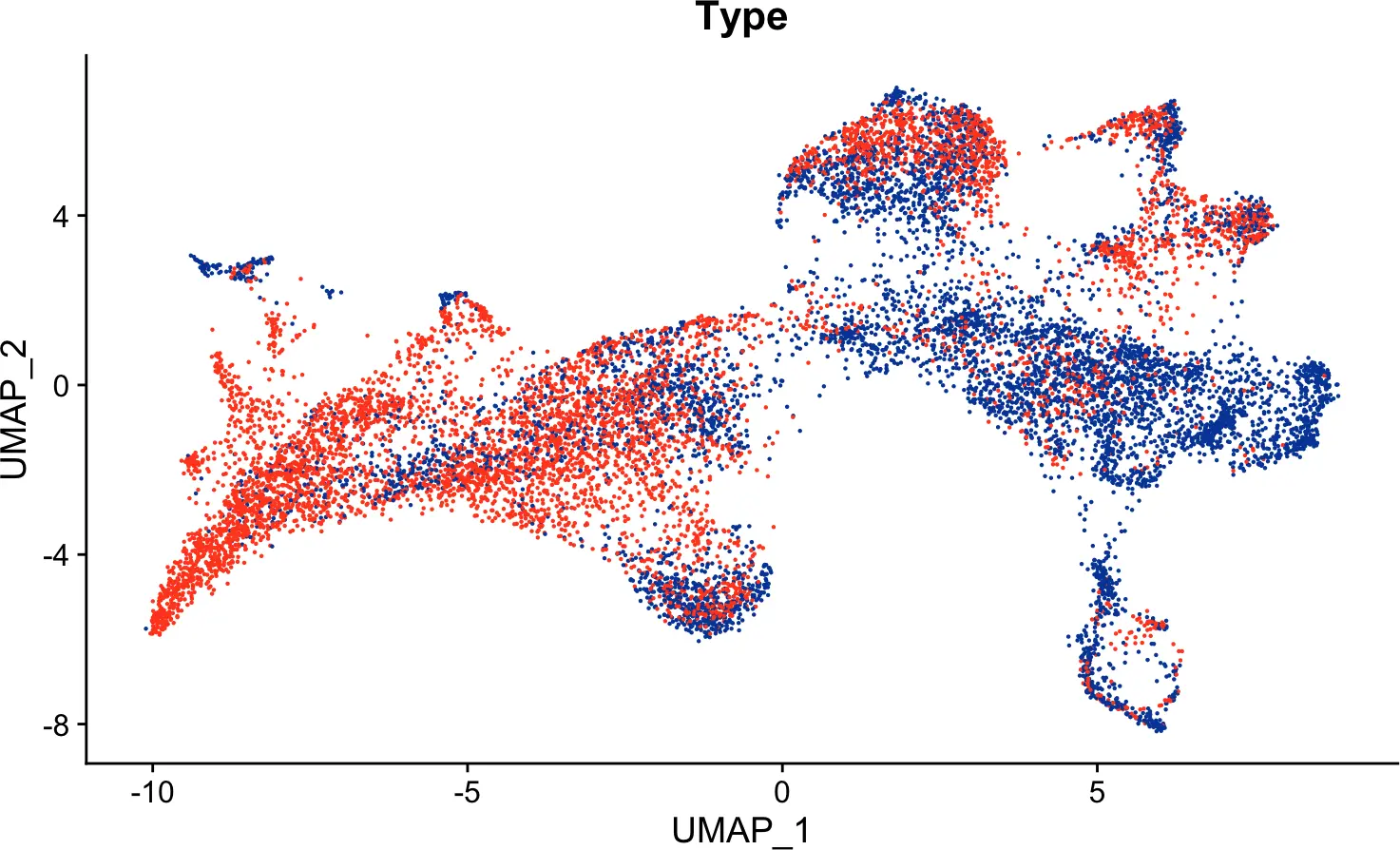
查看外周血和肿瘤浸润T细胞的分布情况。
colorblind_vector <- colorRampPalette(c("#FF4B20", "#FFB433",
"#C6FDEC", "#7AC5FF", "#0348A6"))
DimPlot(seurat, group.by = "Type") + NoLegend() +
scale_color_manual(values=colorblind_vector(2))
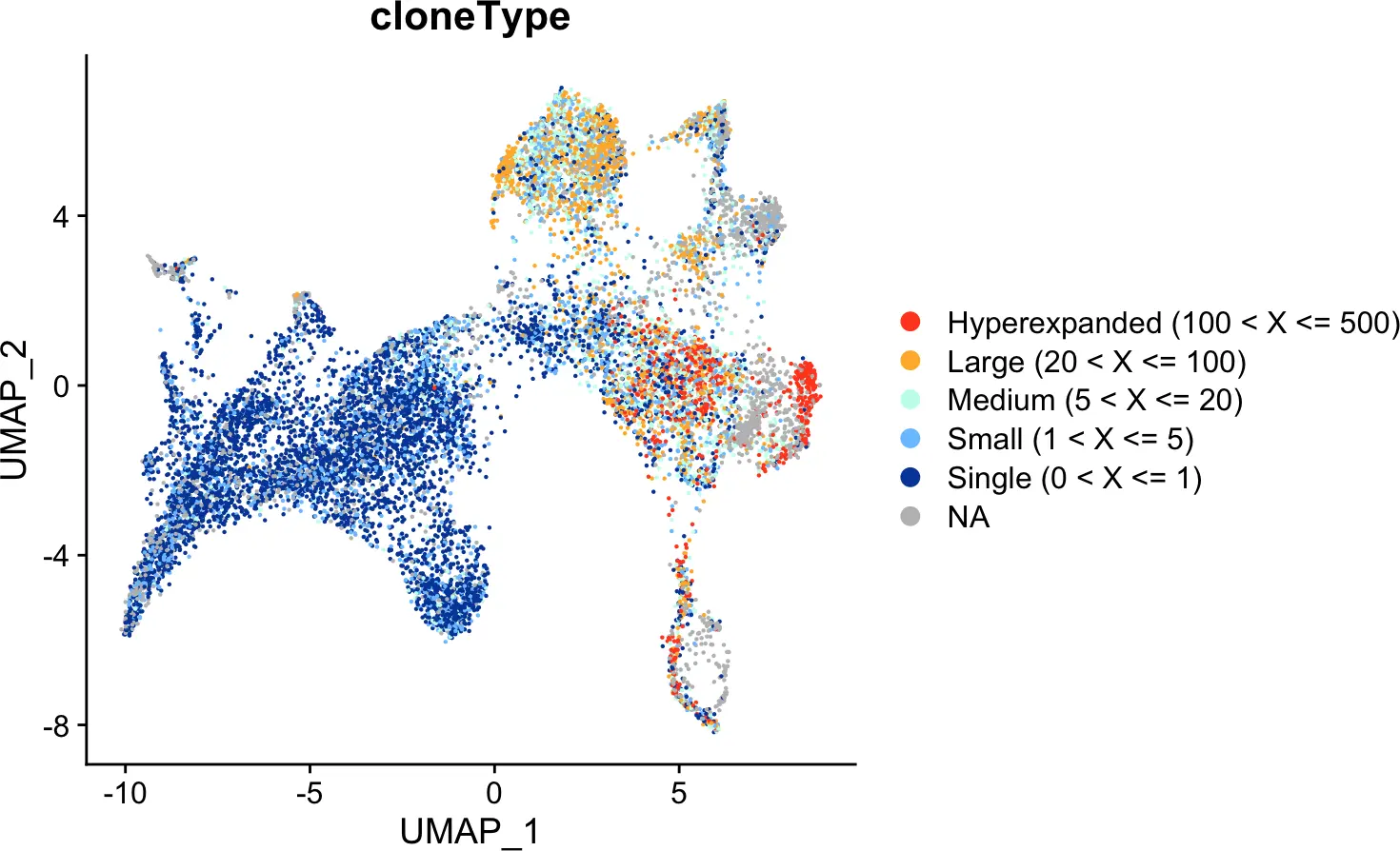
根据clonotype的类型查看其分布情况。
slot(seurat, "meta.data")$cloneType <- factor(slot(seurat, "meta.data")$cloneType,
levels = c("Hyperexpanded (100 < X <= 500)",
"Large (20 < X <= 100)",
"Medium (5 < X <= 20)",
"Small (1 < X <= 5)",
"Single (0 < X <= 1)", NA))
DimPlot(seurat, group.by = "cloneType") +
scale_color_manual(values = colorblind_vector(5), na.value="grey")
6.1 clonalOverlay
我们可以使用clonalOverlay()函数基于降维后的坐标生成克隆扩展细胞位置的叠加情况。
# 分面展示不同样本的情况
clonalOverlay(seurat, reduction = "umap",
freq.cutpoint = 30, bins = 10,
facet = "Patient") +
guides(color = FALSE)
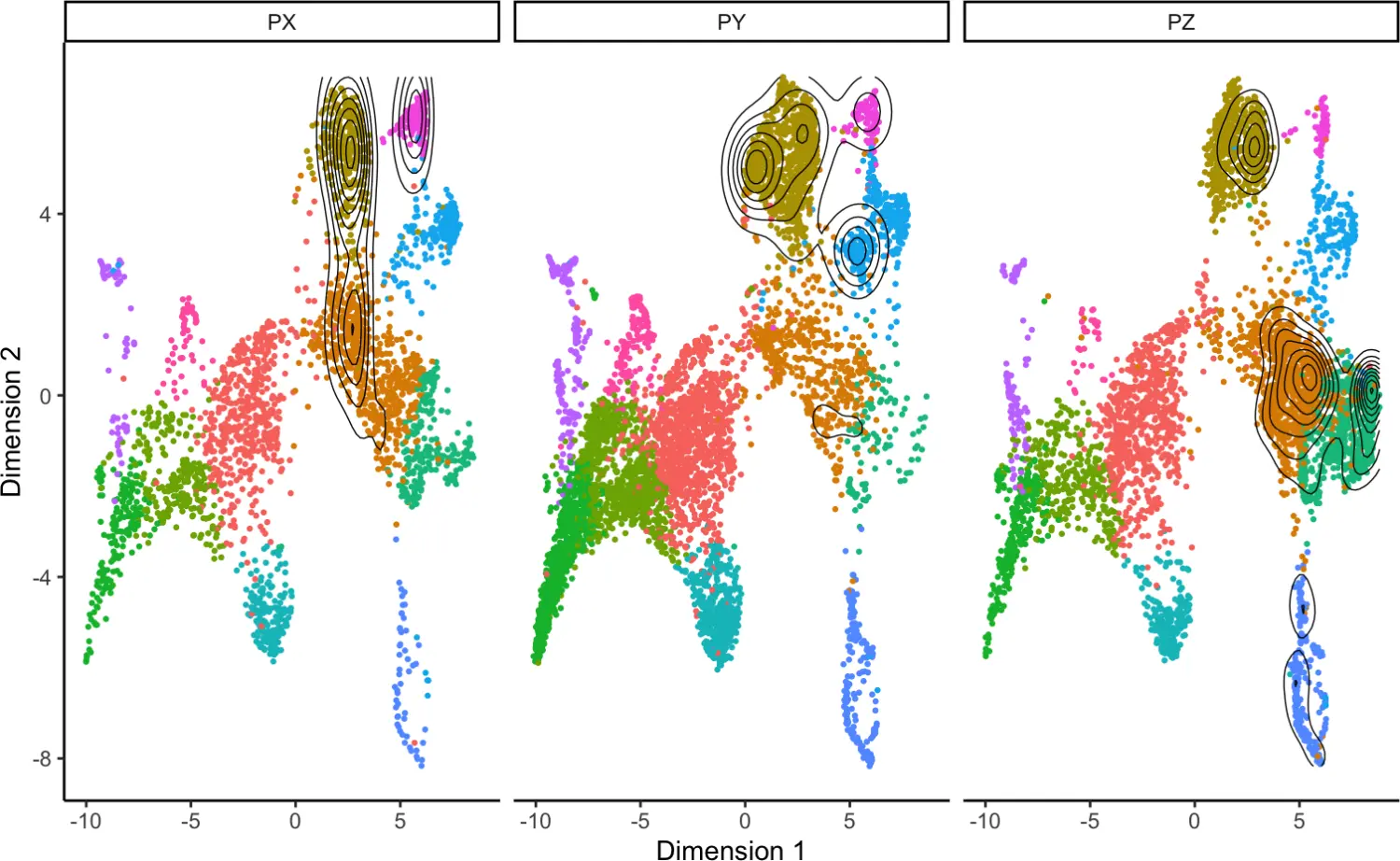
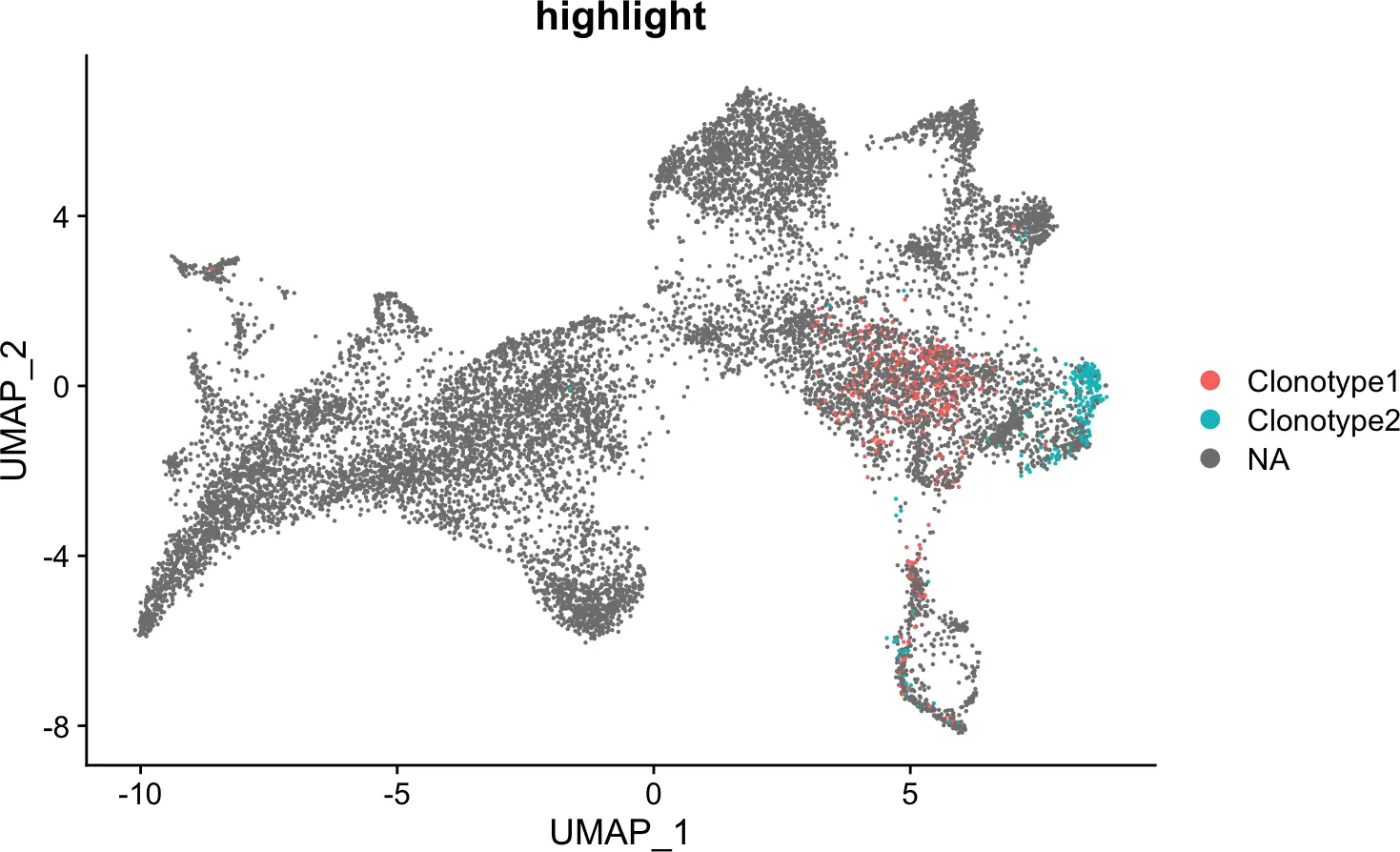
我们还可以使用highlightClonotypes()函数高亮展示特定序列的克隆型分布。
seurat <- highlightClonotypes(seurat, cloneCall= "aa", sequence = c("CAVNGGSQGNLIF_CSAEREDTDTQYF", "NA_CATSATLRVVAEKLFF"))
DimPlot(seurat, group.by = "highlight")
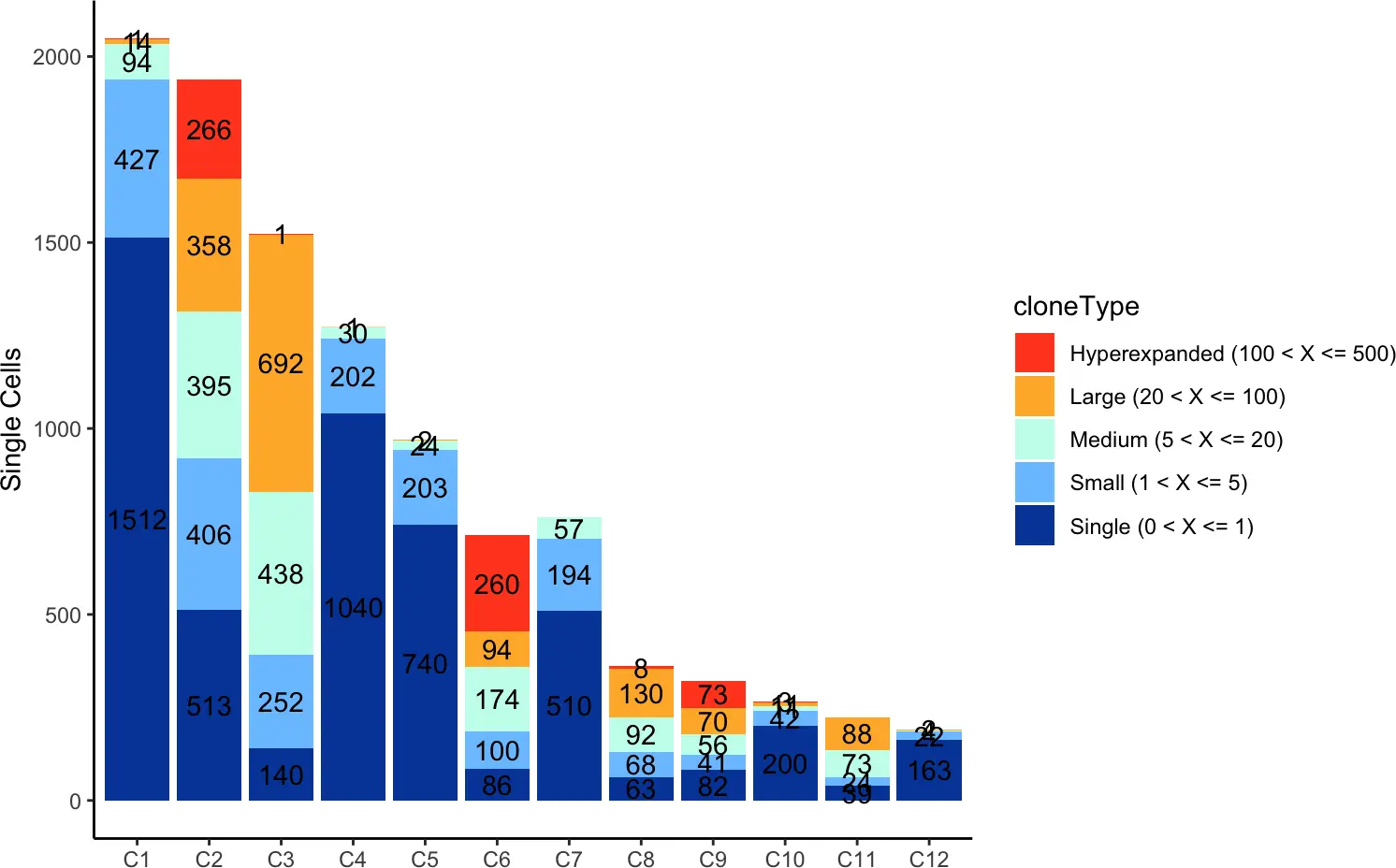
6.2 occupiedscRepertoire
我们可以使用occupiedscRepertoire()函数并通过指定x.axis来展示seurat对象元数据中的cluster或其他变量,从而查看分配到特定频率范围的cluster的细胞数。
occupiedscRepertoire(seurat, x.axis = "cluster")
6.3 alluvialClonotypes
我们可以使用alluvialClonotypes()函数来查看展示跨多个类别的clonotypes。
alluvialClonotypes(seurat, cloneCall = "gene",
y.axes = c("Patient", "cluster", "Type"),
color = "TRAV12-2.TRAJ42.TRAC_TRBV20-1.TRBJ2-3.TRBD2.TRBC2") +
scale_fill_manual(values = c("grey", colorblind_vector(1)))
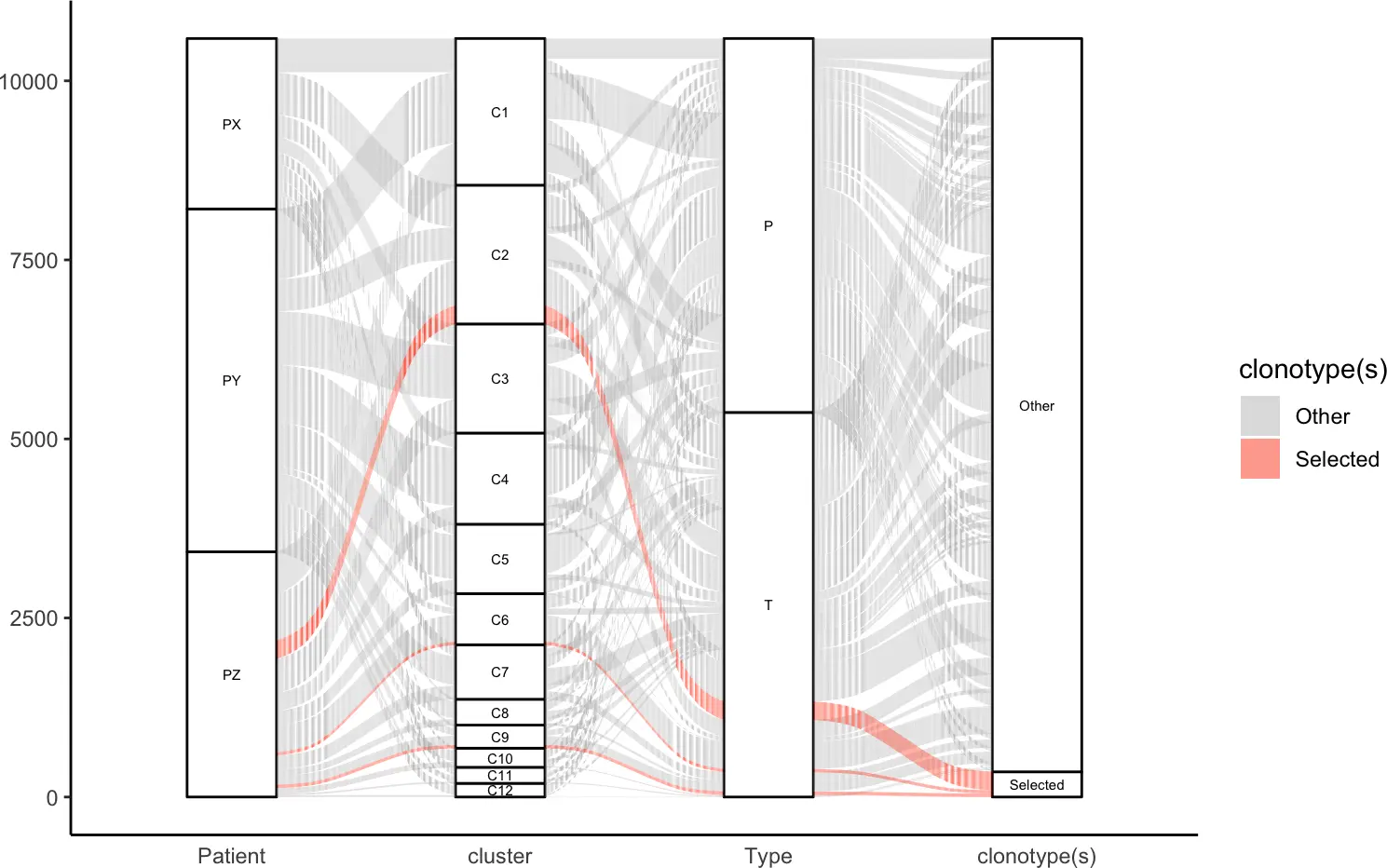
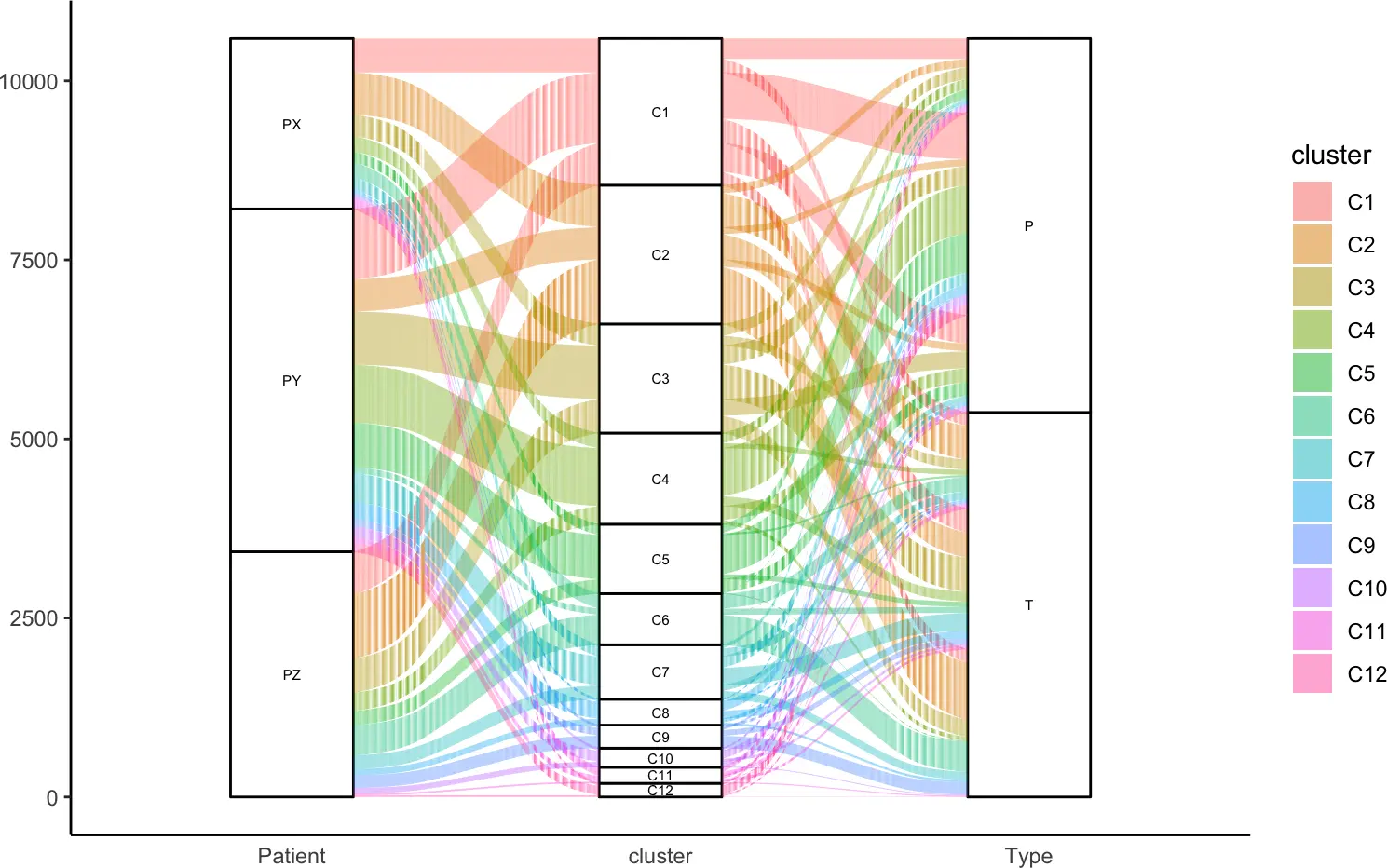
绘制桑基图展示clonotype在不同patient-cluster-type之间的关系。
alluvialClonotypes(seurat, cloneCall = "gene",
y.axes = c("Patient", "cluster", "Type"),
color = "cluster")
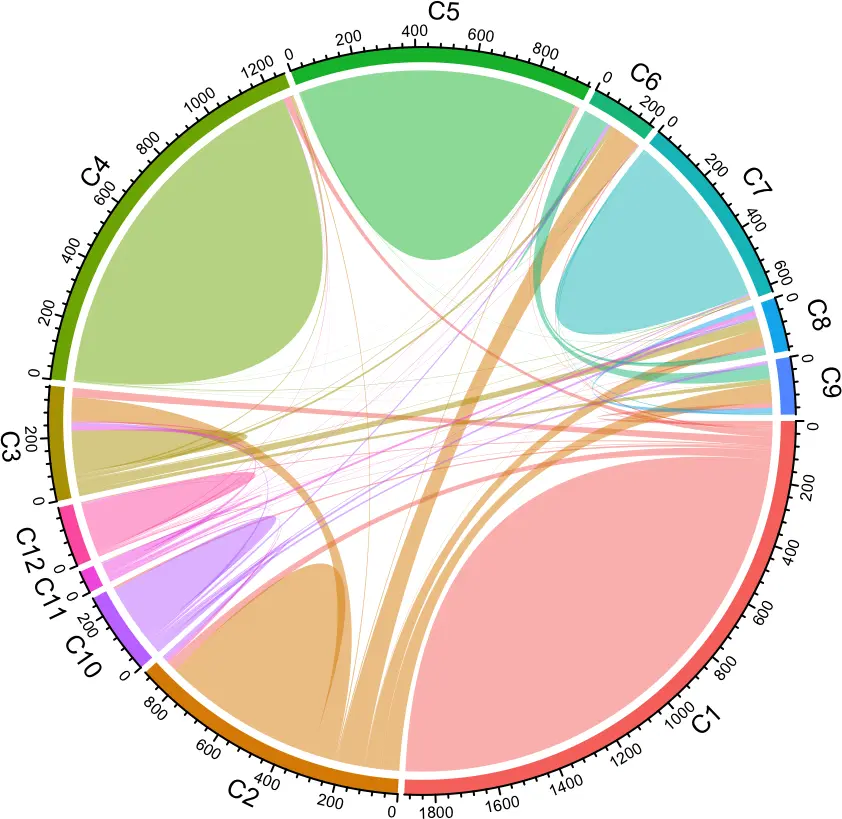
6.4 getCirclize
像桑基图一样,我们也可以使circlize包中的和弦图来可视化cluster之间的互连信息。
library(circlize)
library(scales)
circles <- getCirclize(seurat, groupBy = "cluster")
#Just assigning the normal colors to each cluster
grid.cols <- scales::hue_pal()(length(unique(seurat@active.ident)))
names(grid.cols) <- levels(seurat@active.ident)
#Graphing the chord diagram
circlize::chordDiagram(circles, self.link = 1, grid.col = grid.cols)
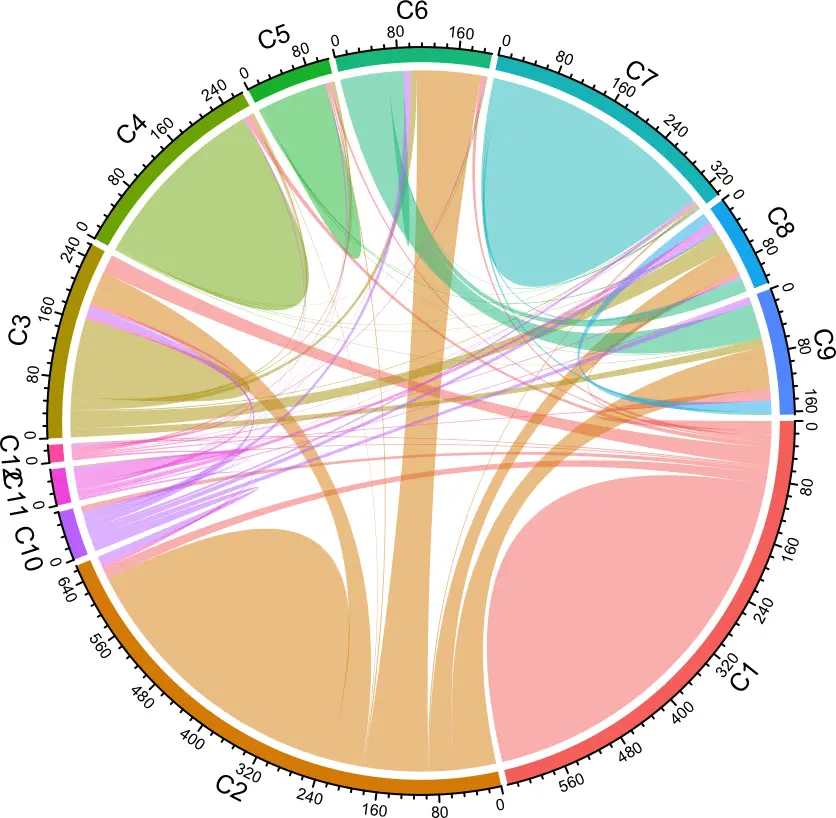
如果我们只想通过对单细胞对象进行子集化来探索肿瘤特异性T细胞,也可以使用此方法。
# 提取子集
subset <- subset(seurat, Type == "T")
circles <- getCirclize(subset, groupBy = "cluster")
grid.cols <- hue_pal()(length(unique(subset@active.ident)))
names(grid.cols) <- levels(subset@active.ident)
chordDiagram(circles, self.link = 1, grid.col = grid.cols)
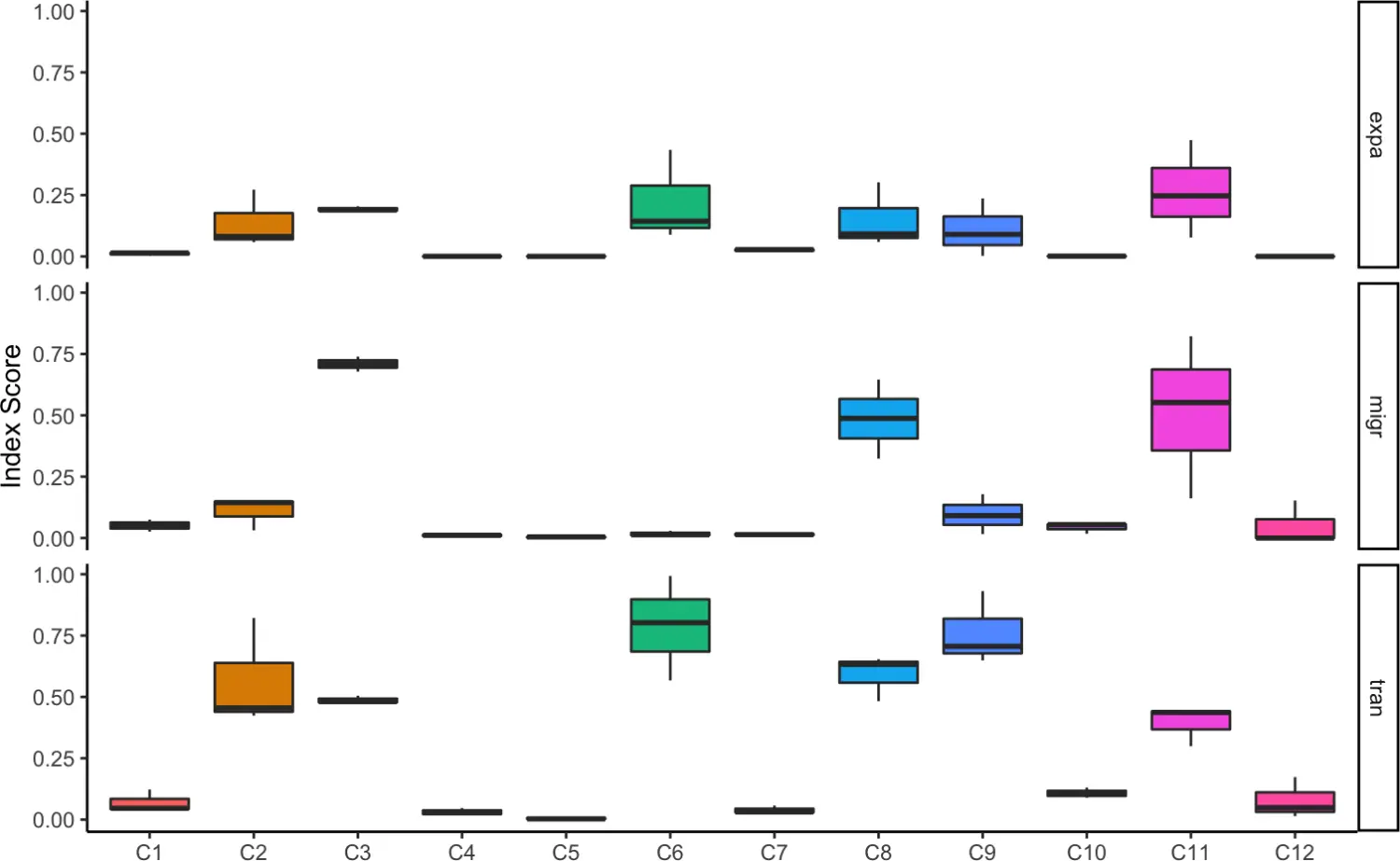
6.5 StartracDiversity
StartracDiversity(seurat, type = "Type",
sample = "Patient", by = "overall")
## [2021-07-08 15:49:51] initialize Startrac ...
## [2021-07-08 15:49:51] calculate startrac index ...
## [2021-07-08 15:49:51] calculate pairwise index ...
## [2021-07-08 15:49:52] calculate indices of each patient ...
## [2021-07-08 15:49:53] collect result
## [2021-07-08 15:49:53] return
7. 可视化细胞聚类后的clonotype
对于希望在Seurat对象中使用元数据来执行scRepertoire提供的分析的用户来说,我们还可以使用expression2List()函数将获取元数据并按cluster将数据输出为列表。
combined2 <- expression2List(seurat, group = "cluster")
length(combined2) #now listed by cluster
## [1] 12
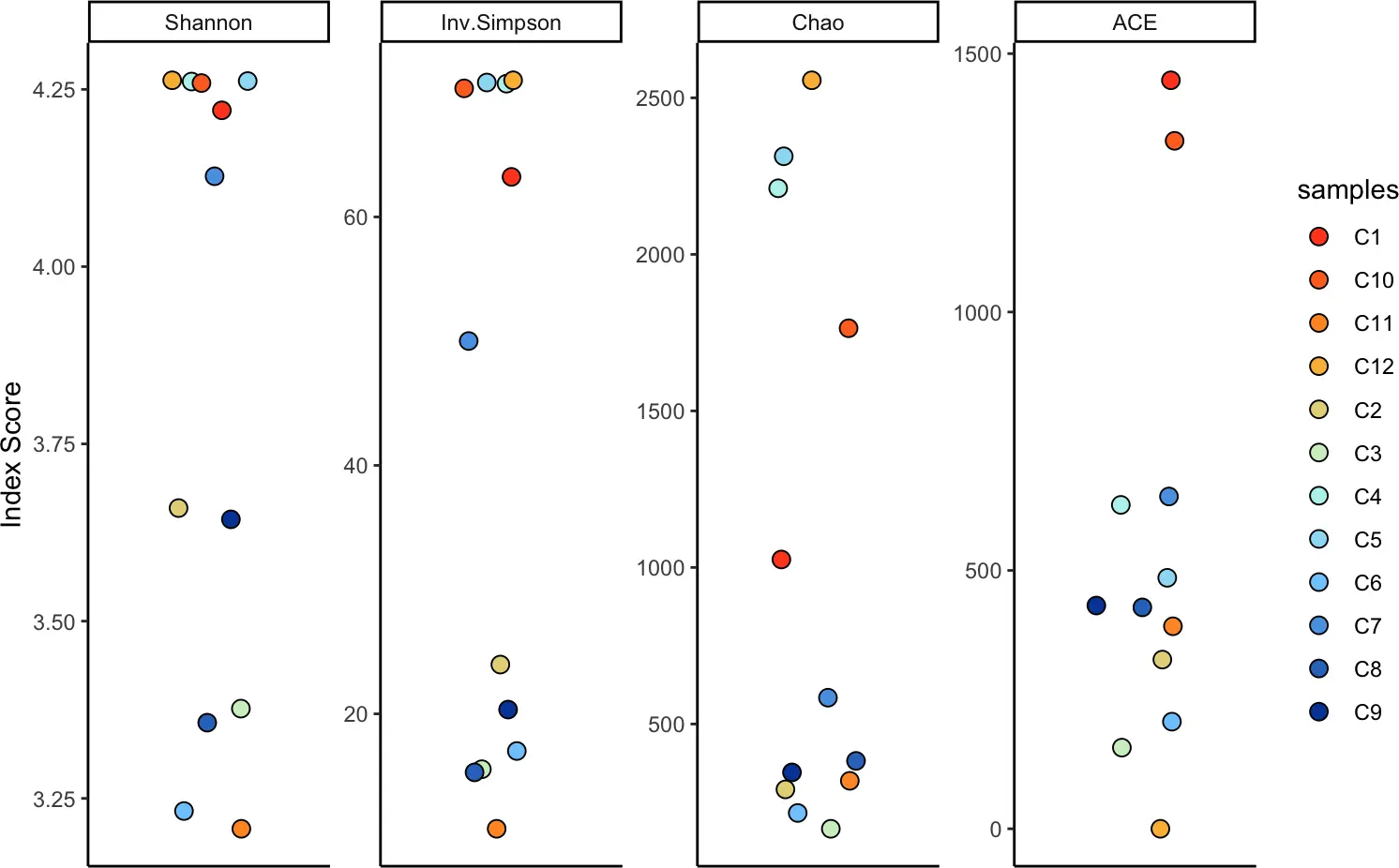
7.1 Clonal Diversity
clonalDiversity(combined2, cloneCall = "nt")
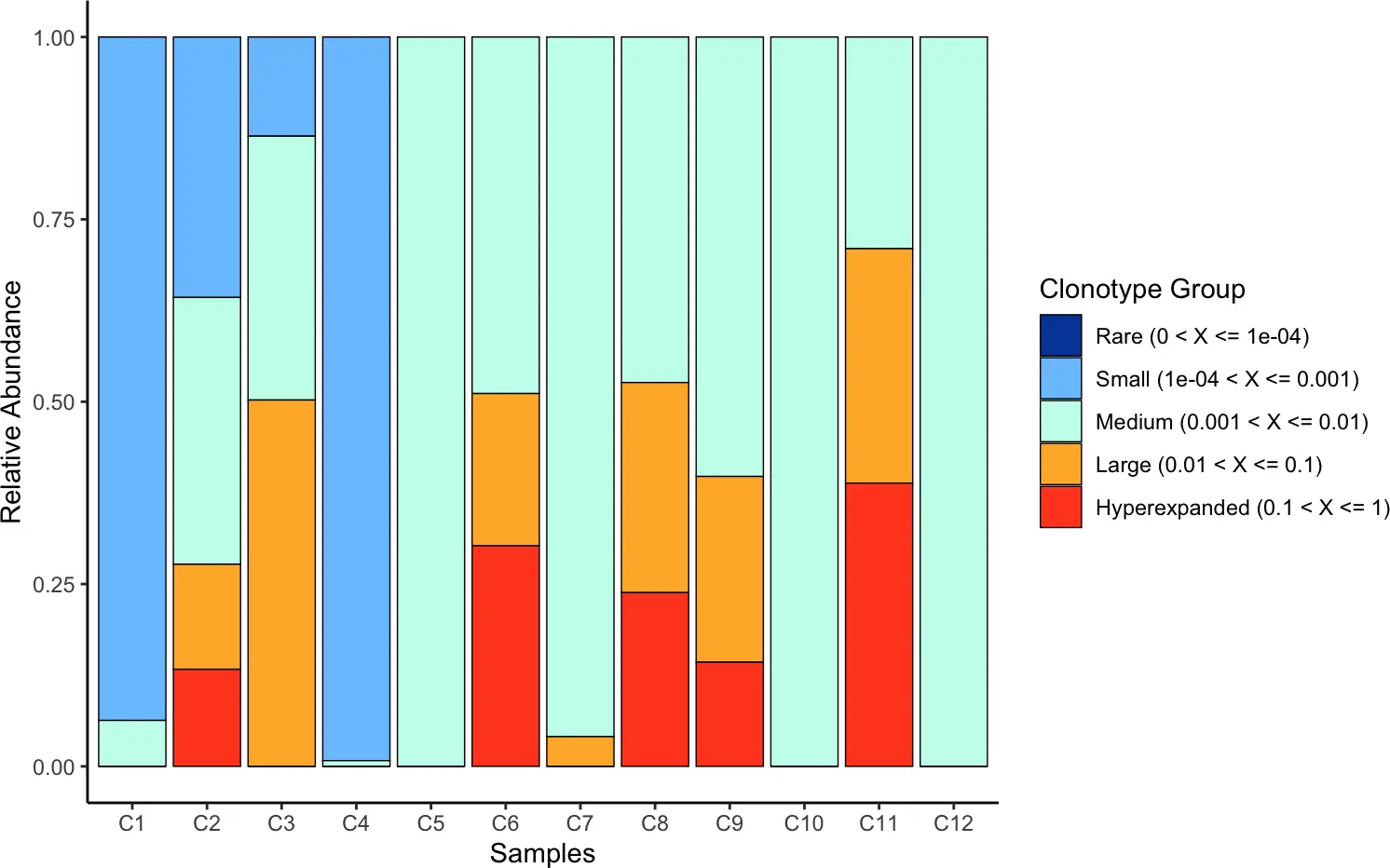
7.2 Clonal Homeostasis
clonalHomeostasis(combined2, cloneCall = "nt")
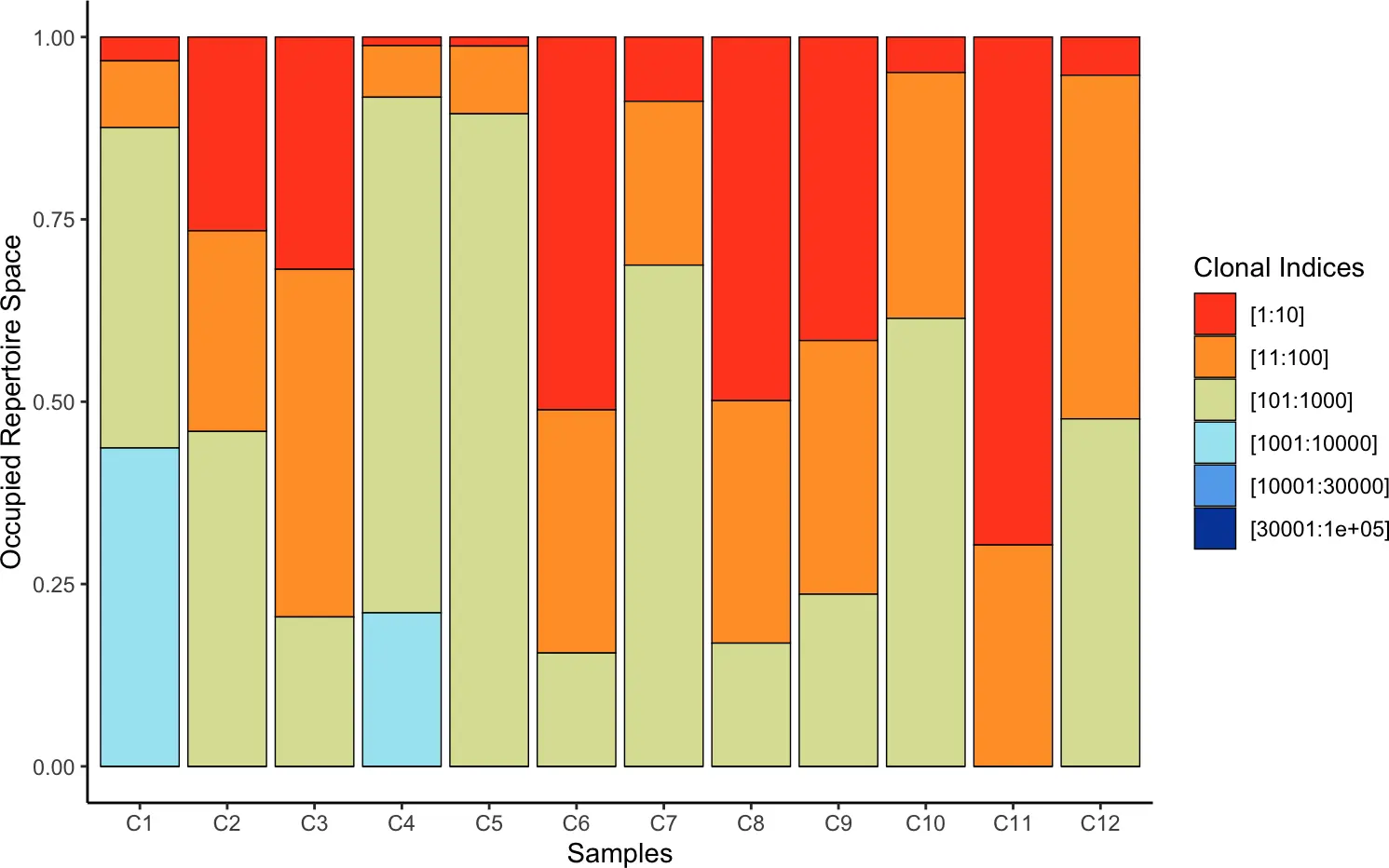
7.3 Clonal Proportion
clonalProportion(combined2, cloneCall = "nt")
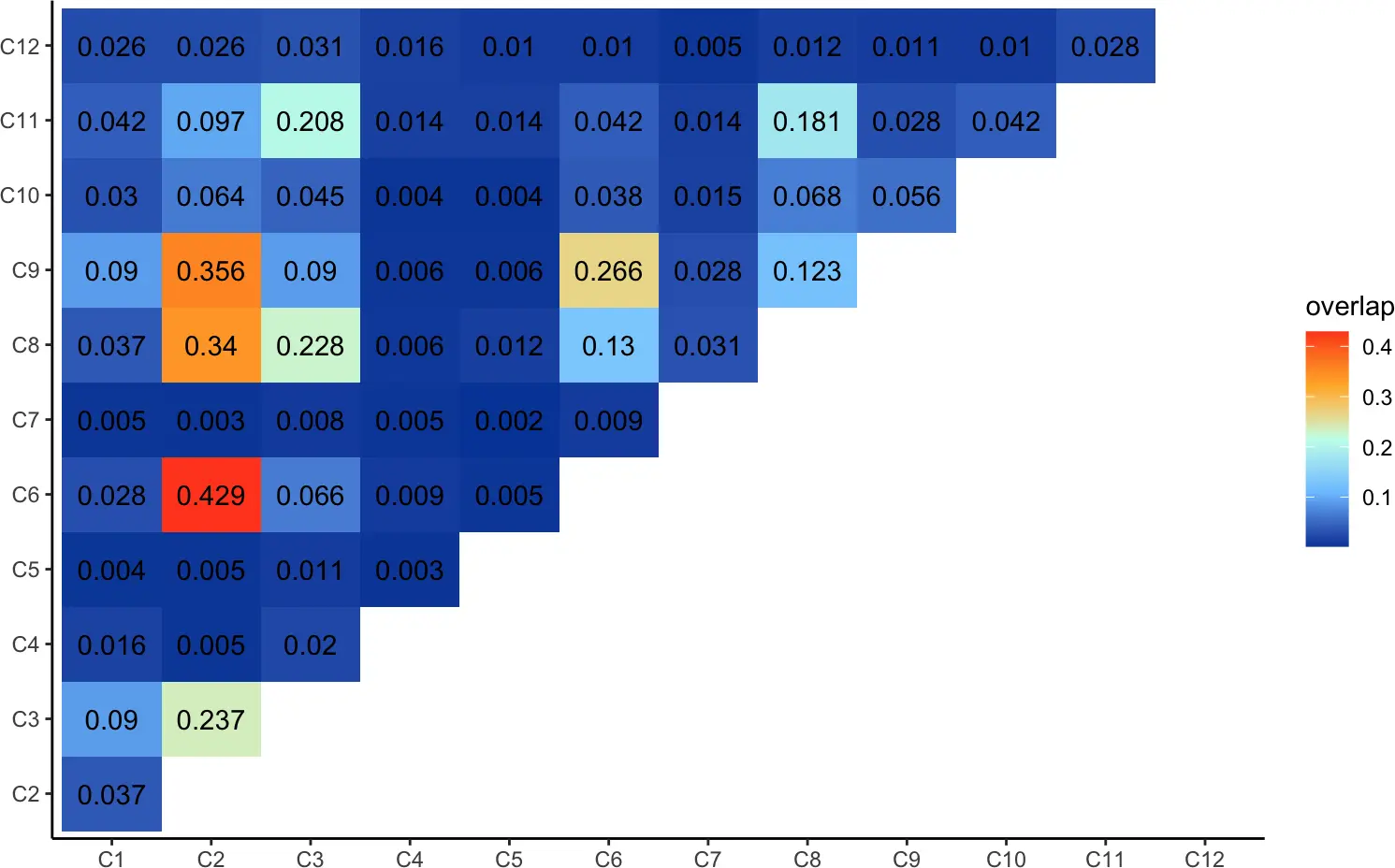
7.4 Clonal Overlap
clonalOverlap(combined2, cloneCall="aa", method="overlap")
参考来源:https://ncborcherding.github.io/vignettes/vignette.html

若有收获,就点个赞吧
0 人点赞
