第七节:空间转录组分析
在本节教程中,我们将学习空间转录组的一些分析方法。10X Visium平台的空间转录组数据在许多方面是类似于scRNA-seq数据的,它包含5-20个细胞而不是单个细胞的UMI计数,但仍然与scRNA-seq相同,数据非常的稀疏,同时还具有相关组织中空间位置的附加信息。
在这里,我们将使用小鼠大脑 (Sagittal)的两个Visium空间转录组数据集进行实战练习。首先,以类似于scRNA-seq数据的方式运行质量控制,然后进行QC过滤,降维,数据整合和细胞聚类。最后,我们使用来自小鼠大脑皮层的scRNA-seq数据运行LabelTransfer来预测Visium空间转录组数据中的细胞类型。
安装并加载所需的R包
# 安装seurat-data包devtools::install_github("satijalab/seurat-data")##checking for file ‘/tmp/RtmpcVJCPY/remotes118eae37e56/satijalab-seurat-data-c633765/DESCRIPTION’ ...✔ checking for file ‘/tmp/RtmpcVJCPY/remotes118eae37e56/satijalab-seurat-data-c633765/DESCRIPTION’ (561ms)##─ preparing ‘SeuratData’:##checking DESCRIPTION meta-information ...✔ checking DESCRIPTION meta-information##─ checking for LF line-endings in source and make files and shell scripts (385ms)##─ checking for empty or unneeded directories##─ building ‘SeuratData_0.2.1.tar.gz’### 加载所需的R包suppressPackageStartupMessages(require(Matrix))suppressPackageStartupMessages(require(dplyr))suppressPackageStartupMessages(require(SeuratData))suppressPackageStartupMessages(require(Seurat))suppressPackageStartupMessages(require(ggplot2))suppressPackageStartupMessages(require(patchwork))suppressPackageStartupMessages(require(dplyr))
加载空间转录组数据集
SeuratData包中包含了几种不同的数据集,其中有来自小鼠大脑和肾脏的空间转录组数据。在这里,我们使用该包中的小鼠大脑数据集进行后续的分析。
outdir = "data/spatial/"
dir.create(outdir, showWarnings = F)
# to list available datasets in SeuratData you can run AvailableData()
# first we dowload the dataset
InstallData("stxBrain")
## Check again that it works, did not work at first...
# now we can list what datasets we have downloaded
InstalledData()
# now we will load the seurat object for one section
brain1 <- LoadData("stxBrain", type = "anterior1")
brain2 <- LoadData("stxBrain", type = "posterior1")
将这两个数据集合并到同一个seuart对象中。
brain <- merge(brain1, brain2)
brain
## An object of class Seurat
## 31053 features across 6049 samples within 1 assay
## Active assay: Spatial (31053 features, 0 variable features)
数据质量控制
同scRNA-seq数据处理一样,我们使用UMI计数(nCount_Spatial),检测到的基因数量(nFeature_Spatial)和线粒体含量等统计指标进行数据质量控制。
# 计算线粒体基因含量
brain <- PercentageFeatureSet(brain, "^mt-", col.name = "percent_mito")
# 计算血红蛋白基因含量
brain <- PercentageFeatureSet(brain, "^Hb.*-", col.name = "percent_hb")
# 使用小提琴图可视化统计指标
VlnPlot(brain, features = c("nCount_Spatial", "nFeature_Spatial", "percent_mito",
"percent_hb"), pt.size = 0.1, ncol = 2) + NoLegend()

我们还可以将以上统计指标绘制到组织切片上。
SpatialFeaturePlot(brain, features = c("nCount_Spatial", "nFeature_Spatial", "percent_mito",
"percent_hb"))
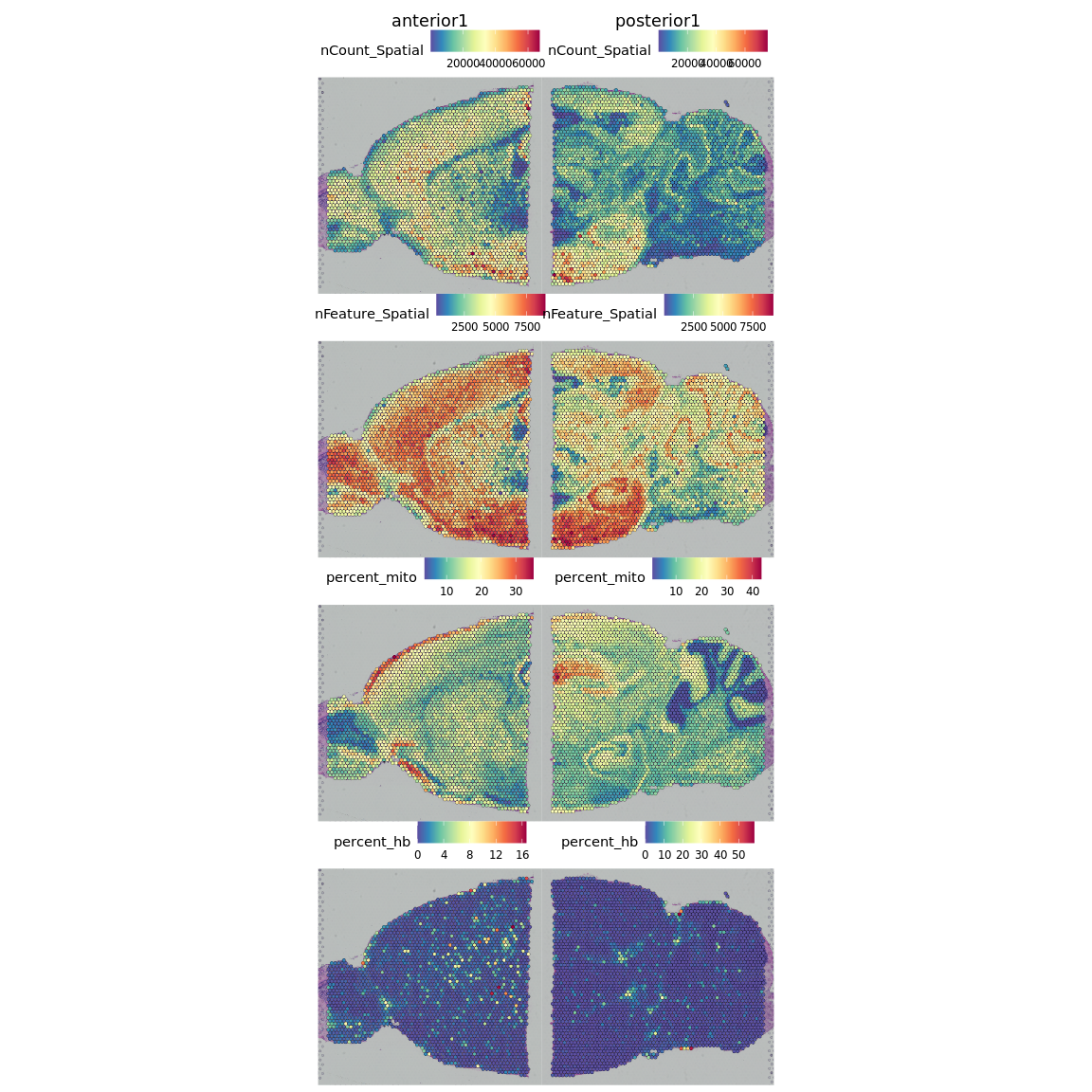
如图所示,counts/features数量少且线粒体基因含量高的spots主要分布于组织的边缘,这些区域很可能是受损的组织。如果您的组织切片中有tears或皱褶,可能还会看到质量较低的组织区域。
但是我们应当注意,对于某些组织类型,表达的基因数目和线粒体的比例也可能是生物学特征,因此请记住您正在处理的组织以及这些特征的含义。
数据过滤筛选
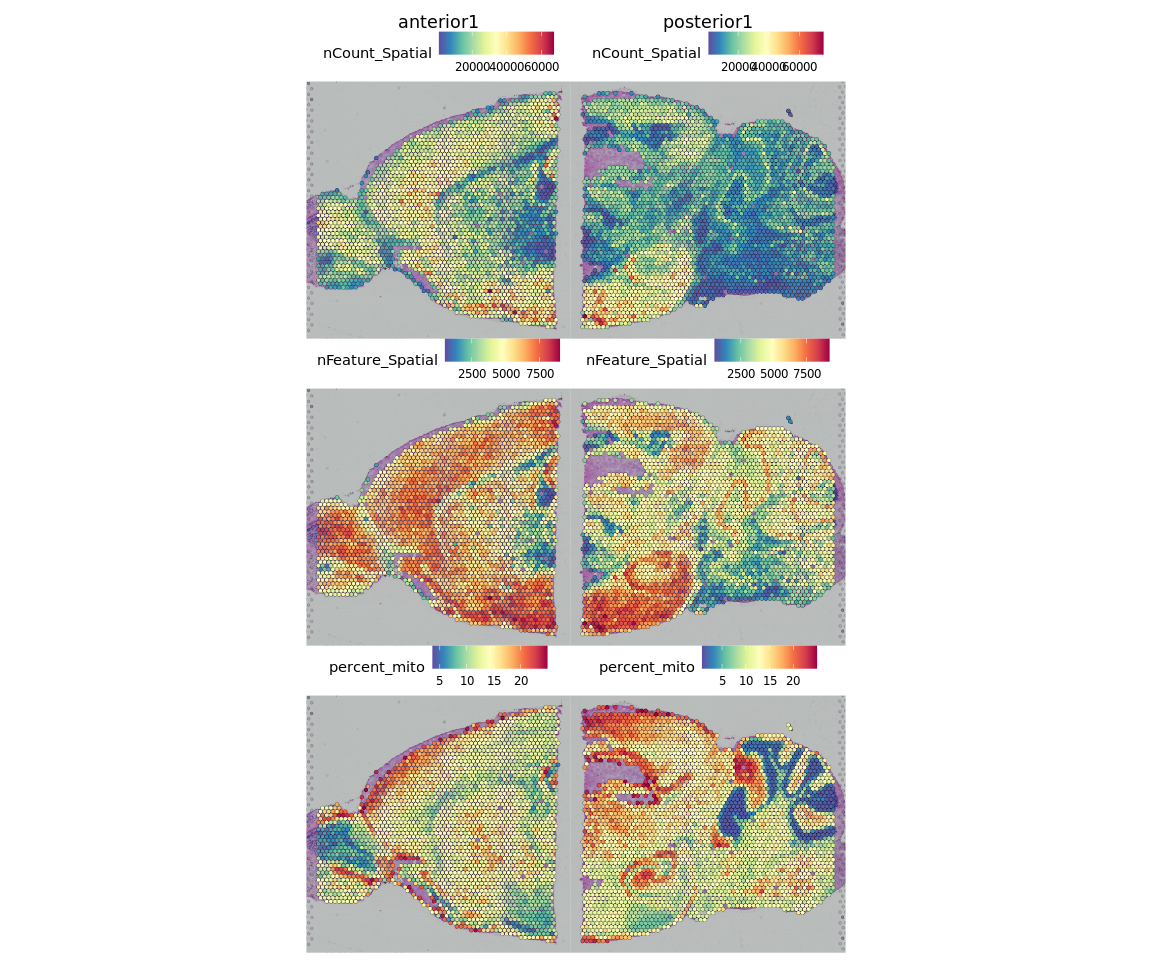
在这里,我们选择线粒体基因含量少于25%,血红蛋白基因含量少于20%和至少检测到的1000个基因的spots。我们必须根据对组织的了解,选择适合自己数据集的过滤标准。
brain = brain[, brain$nFeature_Spatial > 500 & brain$percent_mito < 25 & brain$percent_hb < 20]
并重新可视化过滤后的统计指标:
SpatialFeaturePlot(brain, features = c("nCount_Spatial", "nFeature_Spatial", "percent_mito"))
查看最高表达基因
对于scRNA-seq数据,我们将看看最高表达的基因是哪些。
C = brain@assays$Spatial@counts
C@x = C@x/rep.int(colSums(C), diff(C@p))
most_expressed <- order(Matrix::rowSums(C), decreasing = T)[20:1]
boxplot(as.matrix(t(C[most_expressed, ])), cex = 0.1, las = 1, xlab = "% total count per cell",
col = (scales::hue_pal())(20)[20:1], horizontal = TRUE)
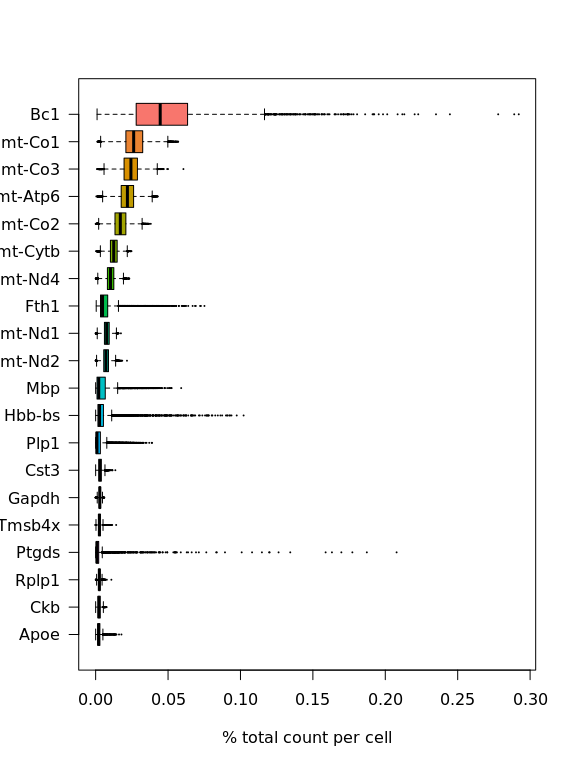
如图所示,线粒体基因是表达最多的基因之一,还有lncRNA基因Bc1(脑细胞质RNA 1)和一种血红蛋白基因。
过滤筛选相关基因
接下来,我们将删除Bc1基因,血红蛋白基因(血液污染)和线粒体基因。
dim(brain)
## [1] 31053 5789
# Filter Bl1
brain <- brain[!grepl("Bc1", rownames(brain)), ]
# Filter Mitocondrial
brain <- brain[!grepl("^mt-", rownames(brain)), ]
# Filter Hemoglobin gene (optional if that is a problem on your data)
brain <- brain[!grepl("^Hb.*-", rownames(brain)), ]
dim(brain)
## [1] 31031 5789
对ST数据进行标准化处理
对于ST数据,Seurat团队推荐使用SCTransfrom方法进行数据标准化,该方法会筛选出高变异的基因,然后对其进行normalization和scaling。
brain <- SCTransform(brain, assay = "Spatial", verbose = TRUE, method = "poisson")
现在,我们可以可视化一些基因的表达。其中,Hpca基因是hippocampal的一个marker,而Ttr是choroid plexus的marker基因
SpatialFeaturePlot(brain, features = c("Hpca", "Ttr"))
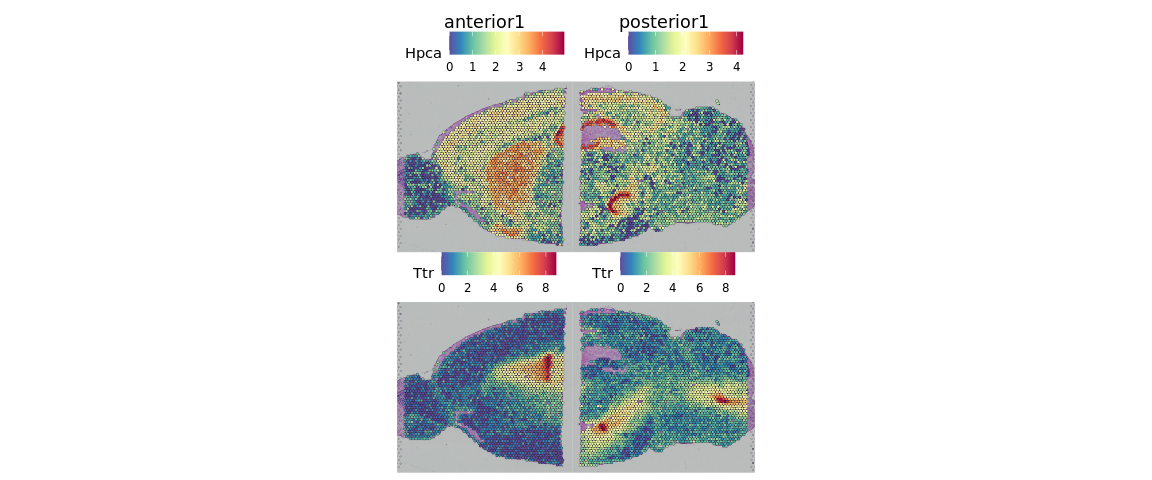
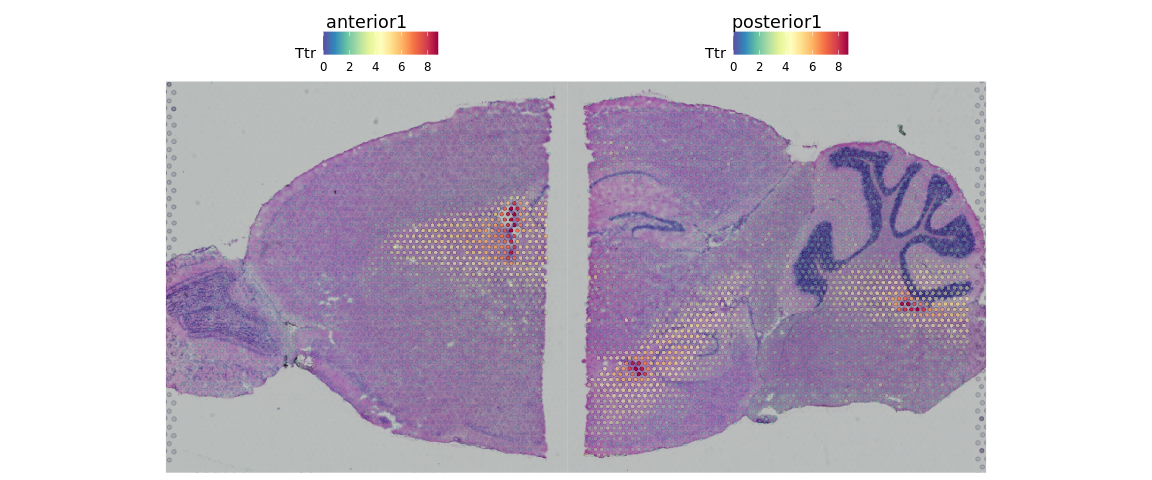
如果想更好的查看这些基因在组织切片上的表达情况,我们还可以调整点的大小和透明度等参数。
SpatialFeaturePlot(brain, features = "Ttr", pt.size.factor = 1, alpha = c(0.1, 1))
降维与聚类
然后,我们可以使用与scRNA-seq数据分析相同的工作流程来进行降维和聚类。
但是,请确保我们是在SCT的assay中进行降维和聚类的。
# PCA线性降维
brain <- RunPCA(brain, assay = "SCT", verbose = FALSE)
# 构建SNN图并进行图聚类
brain <- FindNeighbors(brain, reduction = "pca", dims = 1:30)
brain <- FindClusters(brain, verbose = FALSE)
# UMAP降维可视化
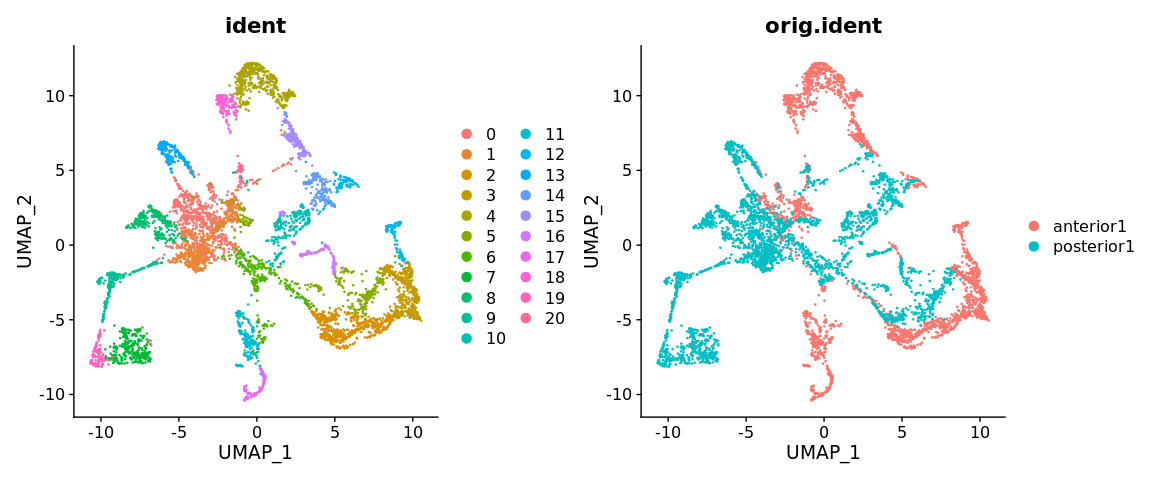
brain <- RunUMAP(brain, reduction = "pca", dims = 1:30)
DimPlot(brain, reduction = "umap", group.by = c("ident", "orig.ident"))
SpatialDimPlot(brain)
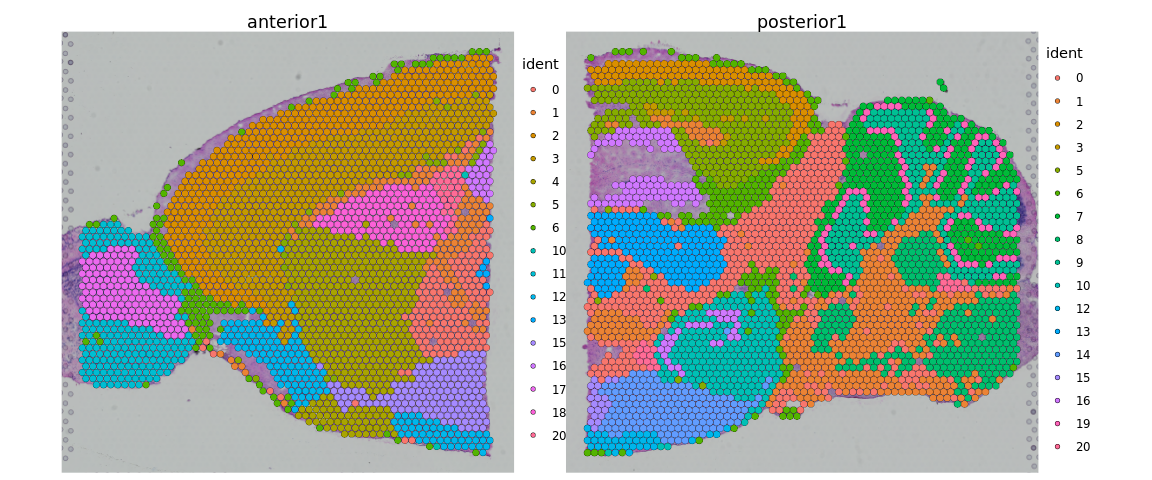
我们还可以分别绘制每个cluster。
SpatialDimPlot(brain, cells.highlight = CellsByIdentities(brain), facet.highlight = TRUE,
ncol = 5)

多样本数据集整合
通常,不同样本的ST数据之间会产生较强的批处理效应,因此最好对其进行数据整合和批次效应的矫正。
在这里,我们将使用SCT分析进行多样本数据集整合。因此,我们需要运行PrepSCTIntegration函数,它将计算两个数据集中所有基因的sctransform残差。
# create a list of the original data that we loaded to start with
st.list = list(anterior1 = brain1, posterior1 = brain2)
# run SCT on both datasets
# 分别对每个数据集进行SCTransform标准化
st.list = lapply(st.list, SCTransform, assay = "Spatial", method = "poisson")
进行多样本数据集整合。
# need to set maxSize for PrepSCTIntegration to work
options(future.globals.maxSize = 2000 * 1024^2) # set allowed size to 2K MiB
# 选择整合的特征基因
st.features = SelectIntegrationFeatures(st.list, nfeatures = 3000, verbose = FALSE)
st.list <- PrepSCTIntegration(object.list = st.list, anchor.features = st.features,
verbose = FALSE)
# 识别整合的anchors
int.anchors <- FindIntegrationAnchors(object.list = st.list, normalization.method = "SCT",
verbose = FALSE, anchor.features = st.features)
# 进行数据整合
brain.integrated <- IntegrateData(anchorset = int.anchors, normalization.method = "SCT",
verbose = FALSE)
rm(int.anchors, st.list)
gc()
## used (Mb) gc trigger (Mb) max used (Mb)
## Ncells 2857164 152.6 4702831 251.2 4702831 251.2
## Vcells 575106704 4387.8 1278197190 9751.9 1065002088 8125.4
然后,像以前一样进行数据降维和聚类。
brain.integrated <- RunPCA(brain.integrated, verbose = FALSE)
brain.integrated <- FindNeighbors(brain.integrated, dims = 1:30)
brain.integrated <- FindClusters(brain.integrated, verbose = FALSE)
brain.integrated <- RunUMAP(brain.integrated, dims = 1:30)
DimPlot(brain.integrated, reduction = "umap", group.by = c("ident", "orig.ident"))
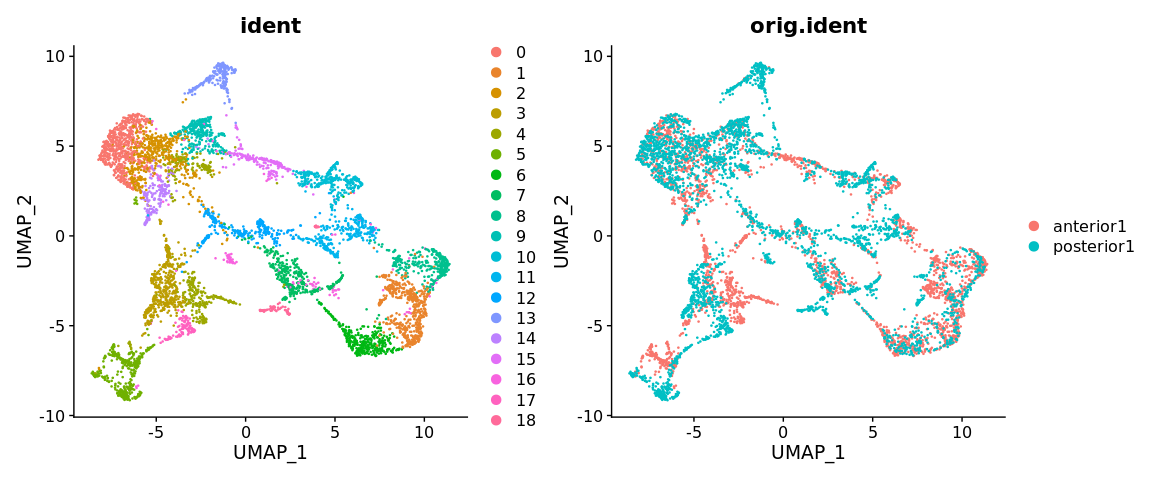
SpatialDimPlot(brain.integrated)

如图所示,你能看出整合后的数据和未整合的数据之间有什么区别吗?你认为哪个分类的效果看起来最好?作为参考,我们可以与Allen brain altlas中的大脑区域进行比较。
识别空间可变基因
目前,有两种计算方法可以识别与组织内的空间位置相关的可变基因。第一种是基于空间上不同的聚类群执行差异表达分析,第二种是在不考虑聚类群或空间注释的情况下查找具有空间特异性的基因。
首先,像scRNA-seq数据那样,我们使用FindMarkers对不同的聚类簇进行差异表达分析。
# differential expression between cluster 1 and cluster 6
de_markers <- FindMarkers(brain.integrated, ident.1 = 5, ident.2 = 6)
# plot top markers
SpatialFeaturePlot(object = brain.integrated, features = rownames(de_markers)[1:3],
alpha = c(0.1, 1), ncol = 3)
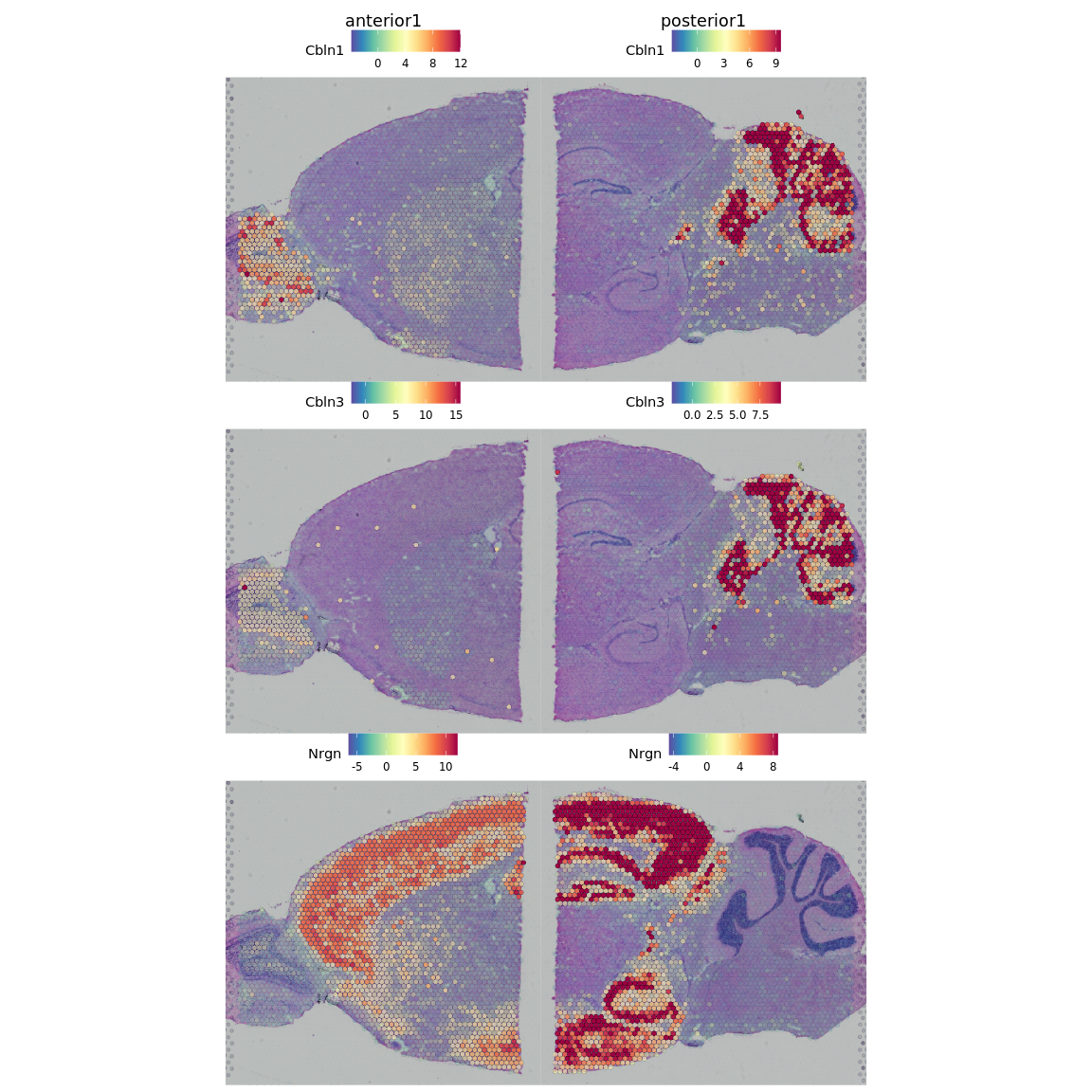
同时,我们还可以使用Seurat包中的FindSpatiallyVariables函数来识别空间可变基因。
brain <- FindSpatiallyVariableFeatures(brain, assay = 'SCT',
features = VariableFeatures(brain)[1:1000],
selection.method = 'markvariogram')
# We would get top features from SpatiallyVariableFeatures
top.features <- head(SpatiallyVariableFeatures(brain, selection.method = 'markvariogram'), 6)
使用scRNA-seq数据集进行整合分析
我们还可以使用scRNA-seq的数据集与空间转录组的数据集进行数据整合分析。这种整合方法非常有用,因为我们可以将scRNA-seq数据中的细胞类型标签转移到Visium空间转录组数据集中。
在这里,我们将使用Allen Institute中产生的约14,000个成年小鼠大脑皮质细胞分类学的scRNA-seq参考数据集进行整合分析,该数据集是通过SMART-Seq2测序技术生成的。
首先,我们使用以下命令将seurat数据从https://www.dropbox.com/s/cuowvm4vrf65pvq/allen_cortex.rds?dl=1下载到文件夹中data/spatial/:
FILE="./data/spatial/allen_cortex.rds"
if [ -e $FILE ]
then
echo "File $FILE is downloaded."
else
echo "Downloading $FILE"
mkdir -p data/spatial
wget -O data/spatial/allen_cortex.rds https://www.dropbox.com/s/cuowvm4vrf65pvq/allen_cortex.rds?dl=1
fi
Subset ST for cortex
由于我们使用的scRNA-seq数据集是由小鼠大脑皮质生成的,因此我们需要将visium数据集中的cortex数据提取出来进行整合分析。
# subset for the anterior dataset
cortex <- subset(brain.integrated, orig.ident == "anterior1")
# there seems to be an error in the subsetting, so the posterior1 image is not
# removed, do it manually
cortex@images$posterior1 = NULL
# subset for a specific region
cortex <- subset(cortex, anterior1_imagerow > 400 | anterior1_imagecol < 150, invert = TRUE)
cortex <- subset(cortex, anterior1_imagerow > 275 & anterior1_imagecol > 370, invert = TRUE)
cortex <- subset(cortex, anterior1_imagerow > 250 & anterior1_imagecol > 440, invert = TRUE)
# also subset for FC clusters
cortex <- subset(cortex, idents = c(0, 1, 6, 7, 12))
p1 <- SpatialDimPlot(cortex, crop = TRUE, label = TRUE)
p2 <- SpatialDimPlot(cortex, crop = FALSE, label = TRUE, pt.size.factor = 1, label.size = 3)
p1 + p2
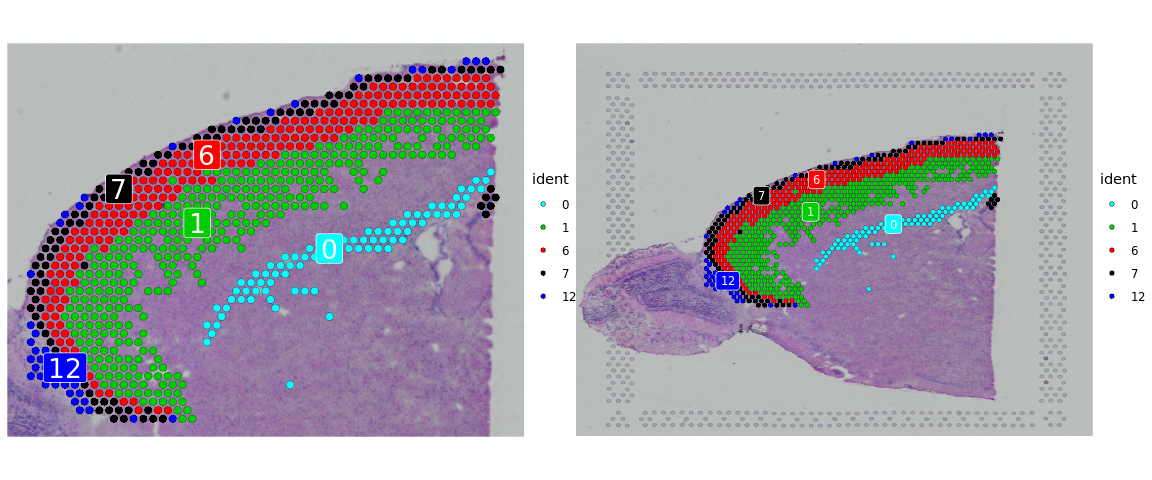
进行数据标准化处理。
# After subsetting, we renormalize cortex
cortex <- SCTransform(cortex, assay = "Spatial", verbose = FALSE, method = "poisson") %>%
RunPCA(verbose = FALSE)
Integrated with scRNA-seq
接下来,我们使用FindTransferAnchors和TransferData函数将空间转录组数据和scRNA-seq数据进行整合。
anchors <- FindTransferAnchors(reference = allen_reference, query = cortex, normalization.method = "SCT")
predictions.assay <- TransferData(anchorset = anchors, refdata = allen_reference$subclass,
prediction.assay = TRUE, weight.reduction = cortex[["pca"]])
cortex[["predictions"]] <- predictions.assay
DefaultAssay(cortex) <- "predictions"
SpatialFeaturePlot(cortex, features = c("L2/3 IT", "L4"), pt.size.factor = 1.6, ncol = 2,
crop = TRUE)
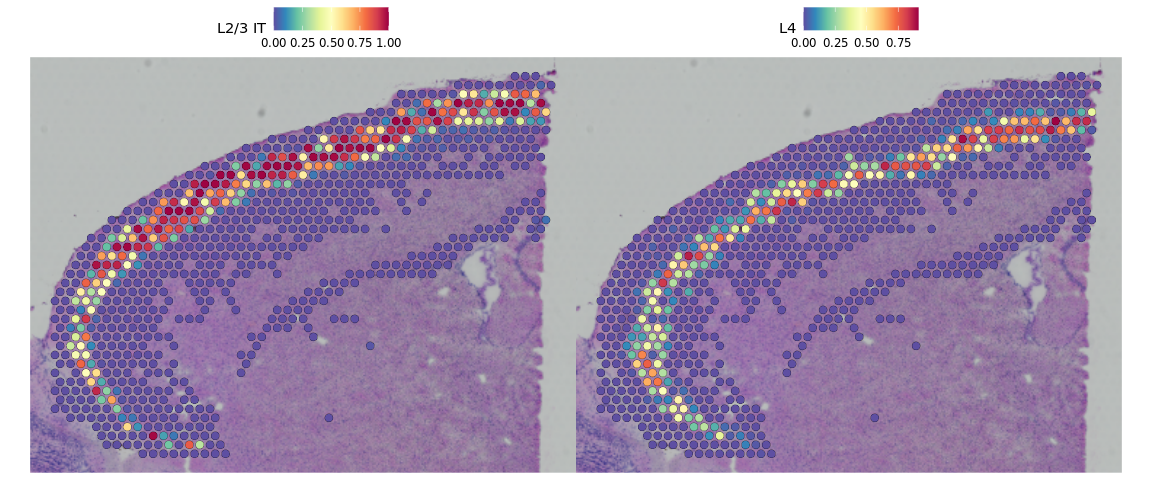
基于这些预测得分,我们可以预测受空间位置限制的特定细胞类型。接下来,我们还可以基于预测出的细胞类型识别空间可变基因。
cortex <- FindSpatiallyVariableFeatures(cortex, assay = "predictions", selection.method = "markvariogram",
features = rownames(cortex), r.metric = 5, slot = "data")
top.clusters <- head(SpatiallyVariableFeatures(cortex), 4)
SpatialPlot(object = cortex, features = top.clusters, ncol = 2)
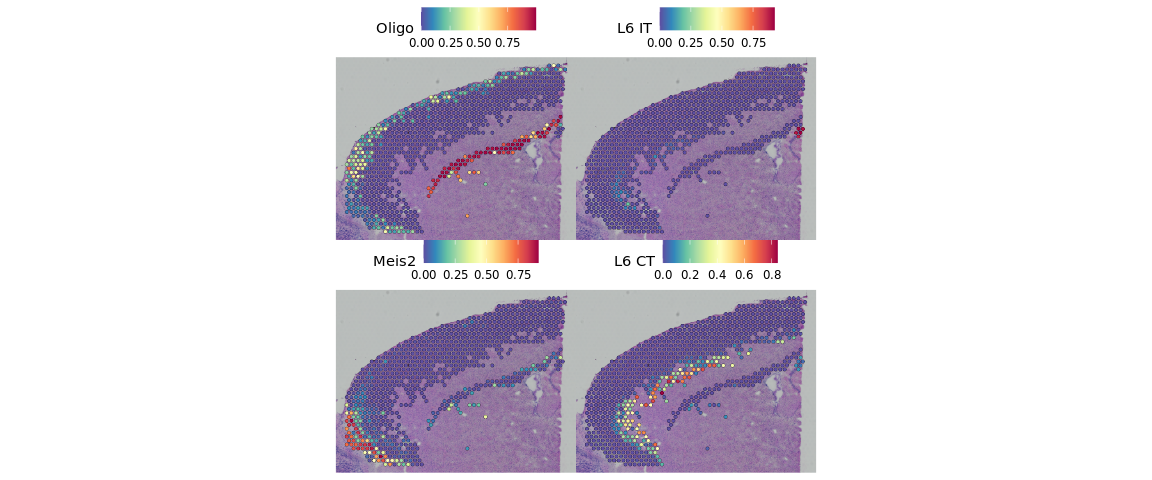
我们还可以在ST数据中查看每个聚类群的预测得分。
VlnPlot(cortex, group.by = "seurat_clusters", features = top.clusters, pt.size = 0,
ncol = 2)
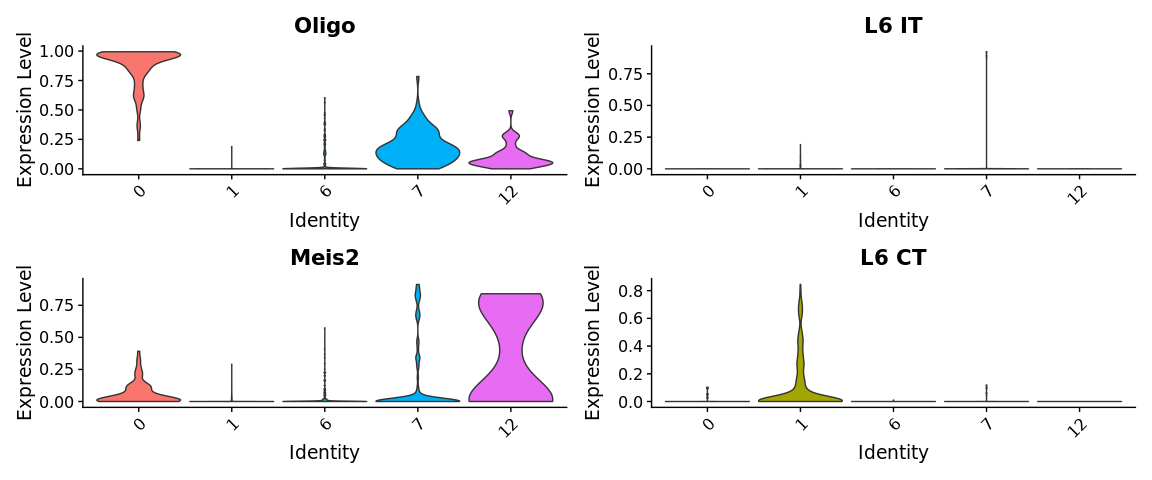
请记住,这些分数仅是预测细胞类型的得分,并不对应于某种细胞类型或相似细胞的比例。它主要告诉我们某个位置的基因表达与特定细胞类型的基因表达高度相似/相异。
如果查看分数,我们会发现某些spots在细胞类型上得到了非常明确的预测,而其他spots在任何一种细胞类型上都没有很高的得分。
sessionInfo()
## R version 3.6.1 (2019-07-05)
## Platform: x86_64-conda_cos6-linux-gnu (64-bit)
## Running under: Ubuntu 20.04 LTS
##
## Matrix products: default
## BLAS/LAPACK: /home/czarnewski/miniconda3/envs/scRNAseq2021/lib/libopenblasp-r0.3.10.so
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] stxBrain.SeuratData_0.1.1 patchwork_1.1.1
## [3] ggplot2_3.3.3 Seurat_3.2.3
## [5] SeuratData_0.2.1 dplyr_1.0.3
## [7] Matrix_1.3-2 RJSONIO_1.3-1.4
## [9] optparse_1.6.6
##
## loaded via a namespace (and not attached):
## [1] Rtsne_0.15 colorspace_2.0-0 deldir_0.2-3
## [4] ellipsis_0.3.1 ggridges_0.5.3 rprojroot_2.0.2
## [7] fs_1.5.0 spatstat.data_1.7-0 farver_2.0.3
## [10] leiden_0.3.6 listenv_0.8.0 remotes_2.2.0
## [13] getopt_1.20.3 ggrepel_0.9.1 RSpectra_0.16-0
## [16] fansi_0.4.2 codetools_0.2-18 splines_3.6.1
## [19] knitr_1.30 polyclip_1.10-0 pkgload_1.1.0
## [22] jsonlite_1.7.2 ica_1.0-2 cluster_2.1.0
## [25] png_0.1-7 uwot_0.1.10 sctransform_0.3.2
## [28] shiny_1.5.0 compiler_3.6.1 httr_1.4.2
## [31] assertthat_0.2.1 fastmap_1.0.1 lazyeval_0.2.2
## [34] limma_3.42.0 cli_2.2.0 later_1.1.0.1
## [37] formatR_1.7 htmltools_0.5.1 prettyunits_1.1.1
## [40] tools_3.6.1 rsvd_1.0.3 igraph_1.2.6
## [43] gtable_0.3.0 glue_1.4.2 reshape2_1.4.4
## [46] RANN_2.6.1 rappdirs_0.3.1 spatstat_1.64-1
## [49] Rcpp_1.0.6 scattermore_0.7 vctrs_0.3.6
## [52] nlme_3.1-150 lmtest_0.9-38 xfun_0.20
## [55] stringr_1.4.0 globals_0.14.0 ps_1.5.0
## [58] testthat_3.0.1 mime_0.9 miniUI_0.1.1.1
## [61] lifecycle_0.2.0 irlba_2.3.3 devtools_2.3.2
## [64] goftest_1.2-2 future_1.21.0 MASS_7.3-53
## [67] zoo_1.8-8 scales_1.1.1 spatstat.utils_1.20-2
## [70] promises_1.1.1 parallel_3.6.1 RColorBrewer_1.1-2
## [73] yaml_2.2.1 curl_4.3 gridExtra_2.3
## [76] memoise_1.1.0 reticulate_1.18 pbapply_1.4-3
## [79] rpart_4.1-15 stringi_1.5.3 desc_1.2.0
## [82] pkgbuild_1.2.0 rlang_0.4.10 pkgconfig_2.0.3
## [85] matrixStats_0.57.0 evaluate_0.14 lattice_0.20-41
## [88] tensor_1.5 ROCR_1.0-11 purrr_0.3.4
## [91] labeling_0.4.2 htmlwidgets_1.5.3 cowplot_1.1.1
## [94] processx_3.4.5 tidyselect_1.1.0 parallelly_1.23.0
## [97] RcppAnnoy_0.0.18 plyr_1.8.6 magrittr_2.0.1
## [100] R6_2.5.0 generics_0.1.0 DBI_1.1.1
## [103] mgcv_1.8-33 pillar_1.4.7 withr_2.4.0
## [106] fitdistrplus_1.1-3 abind_1.4-5 survival_3.2-7
## [109] tibble_3.0.5 future.apply_1.7.0 crayon_1.3.4
## [112] KernSmooth_2.23-18 plotly_4.9.3 rmarkdown_2.6
## [115] usethis_2.0.0 grid_3.6.1 data.table_1.13.6
## [118] callr_3.5.1 digest_0.6.27 xtable_1.8-4
## [121] tidyr_1.1.2 httpuv_1.5.5 munsell_0.5.0
## [124] viridisLite_0.3.0 sessioninfo_1.1.1
参考来源:https://nbisweden.github.io/workshop-scRNAseq/labs/compiled/seurat/seurat_07_spatial.html

若有收获,就点个赞吧
0 人点赞
