本文来自“单细胞组学”公众号 原文链接:https://mp.weixin.qq.com/s/jUw79trMDRSAwCphSIbweA
ATAC-seq即transposase-accessible chromatin using sequencing,是一种检测染色体开放区域的技术。该方法由斯坦福大学Greenleaf实验室在2013年首次发表[1],两年后该实验室又发表基于单细胞的ATAC-seq技术[2]。
ATAC-seq相比于之前的FAIRE-seq和DNase-seq,ATAC-seq用Tn5转座酶对染色体开放区域进行剪切,并加上adaptors序列(图1的红蓝片段)。最后得到的DNA片段,包括了开放区域的剪切片段,以及横跨一个或多个核小体的长片段。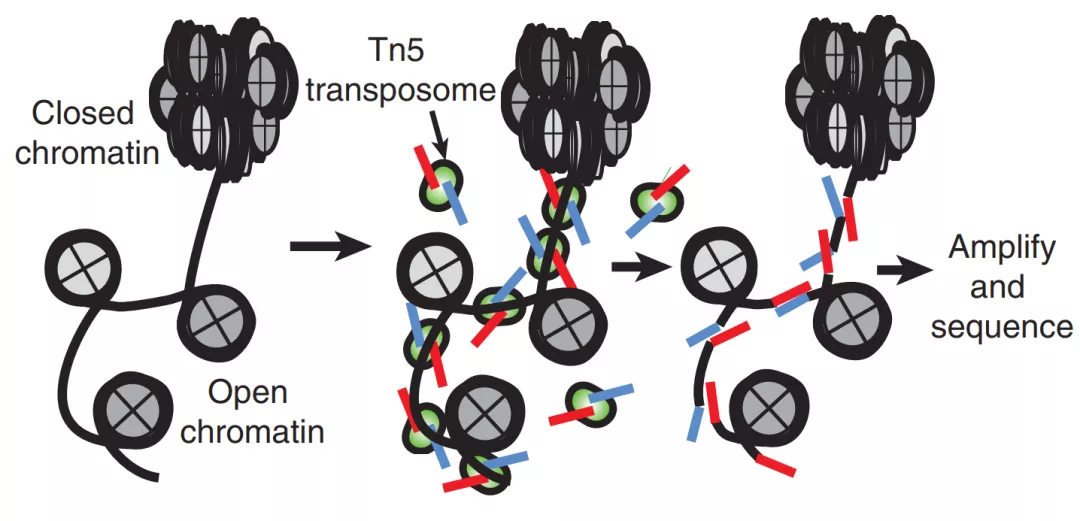
图1. ATAC-seq[1]
根据片段长度可以分为Fragments in nucleosome-free regions(<147 base pairs)和**Fragments flanking a single nucleosome** (147~294 base pairs), 以及更长的多核片段。片段长度分布如下图,**没有跨越核小体的小片段最多**,其次是单核片段,依次递减。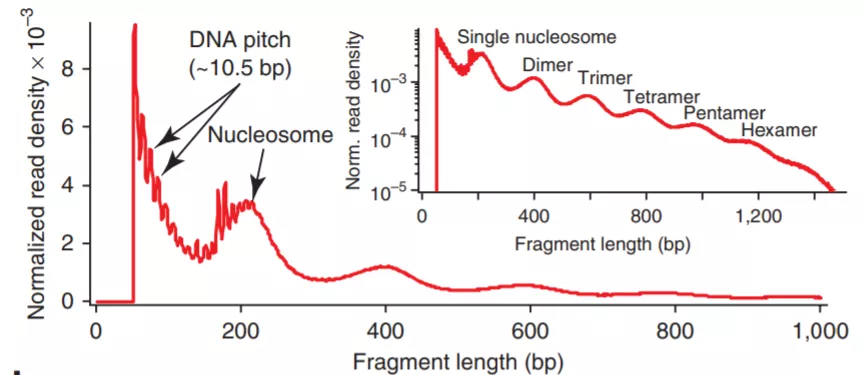
图2. DNA片段长度分布
scATAC-seq建库原理
以10x 建库方法为例[5],比较scATAC-seq 和scRNA-seq建库方法的异同。
二者都用胶珠(GEMs)的方法,不一样的是ATAC胶珠上的序列中不用UMI,因为基因组只有一对序列,无需像RNA一样定量。另外序列末端用接头引物Read 1N代替PolyT。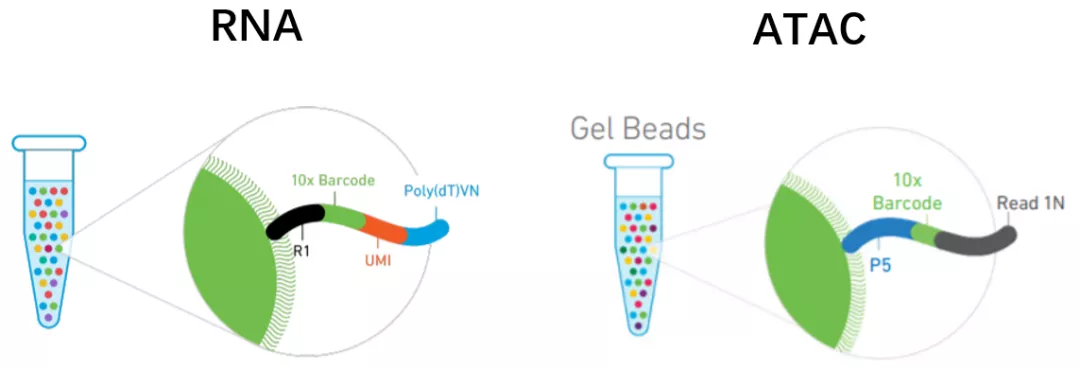
scRNA-seq通过结合cDNA的PolyA尾进行扩增,而scATAC-seq的DNA片段没有PolyA尾,取而代之的是Tn5酶转座剪切时插入的adaptors片段,可以与胶珠上的Read 1N序列互补。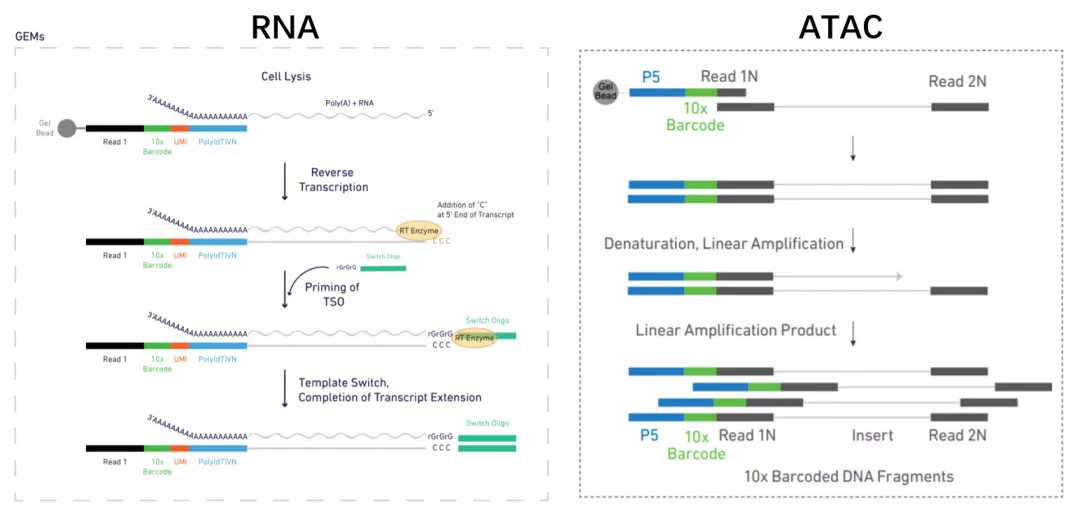
DNA片段接上胶珠后,在另一端加Read2和Sample index序列。在此之前,scRNA-seq需要将cDNA酶切至合适的片段长度,而scATAC-seq的片段不进行打碎,接上Sample index和P7序列后进行扩增。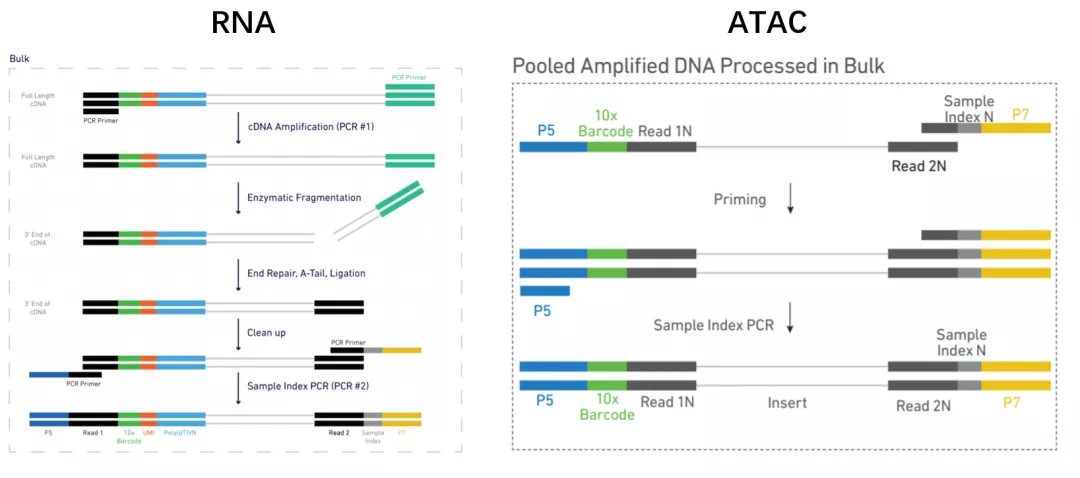
最后上机测序。scRNAseq如果是3‘单端测序,Read2读取最近的100bp读长,而Read1只读取16bp的细胞barcode序列和10bp的UMI序列,共26bp。scATAC-seq则用双末端测序,读长一般不低于45bp[3]。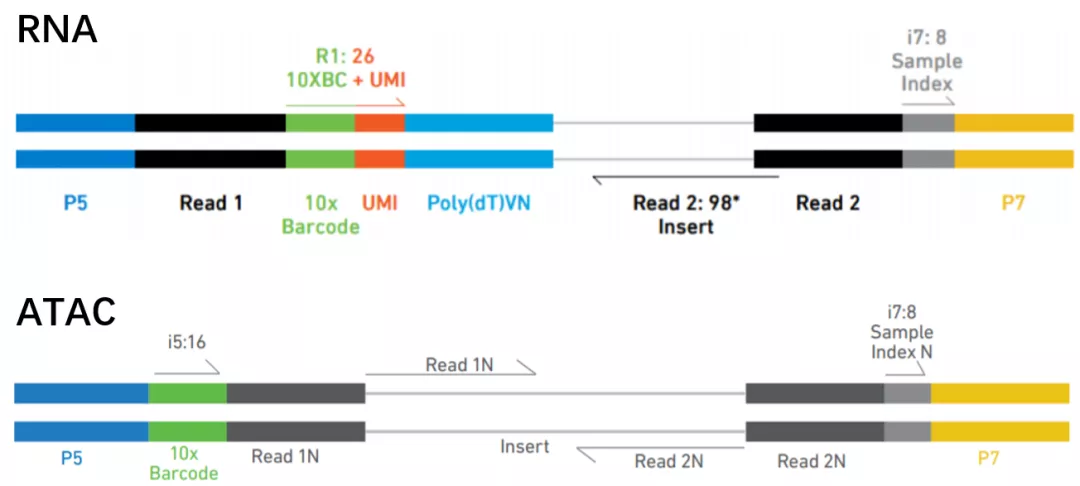
scATAC-seq最后可以得到4个原始文件:
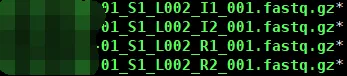
其中I1/2分别是barcode和sample index,R1/2是目的片段的双末端。
10x提供cellranger软件对原始数据进行初步分析,如质控,比对,peak calling等。
$ cellranger-atac count --id=sample345 \--reference=/opt/refdata-cellranger-atac-GRCh38-1.2.0 \--fastqs=/home/jdoe/runs/HAWT7ADXX/outs/fastq_path \--sample=mysample \--localcores=8 \--localmem=64
质控方法
重要指标:
1. Fraction of fragments overlapping any targeted region
覆盖注释区域的片段比例。目标区域包括TSS,DNase HS,增强子和启动子等。一般要求大于55%。
2. Fraction of transposition events in peaks in cell barcodes
转座位点位于peaks区域的比例。一般大于25%。如果比例过小,有可能是细胞状态不好,或者测序深度不够。
Enrichment score of transcription start sites(TSS)
转录起始位点的标准化分数。阈值选择根据基因组而定。人类一般选择TSS分数大于5或者6的细胞样本[3]。如果分数太低说明细胞染色质结构瓦解,或者实验过程细胞裂解方式不当。
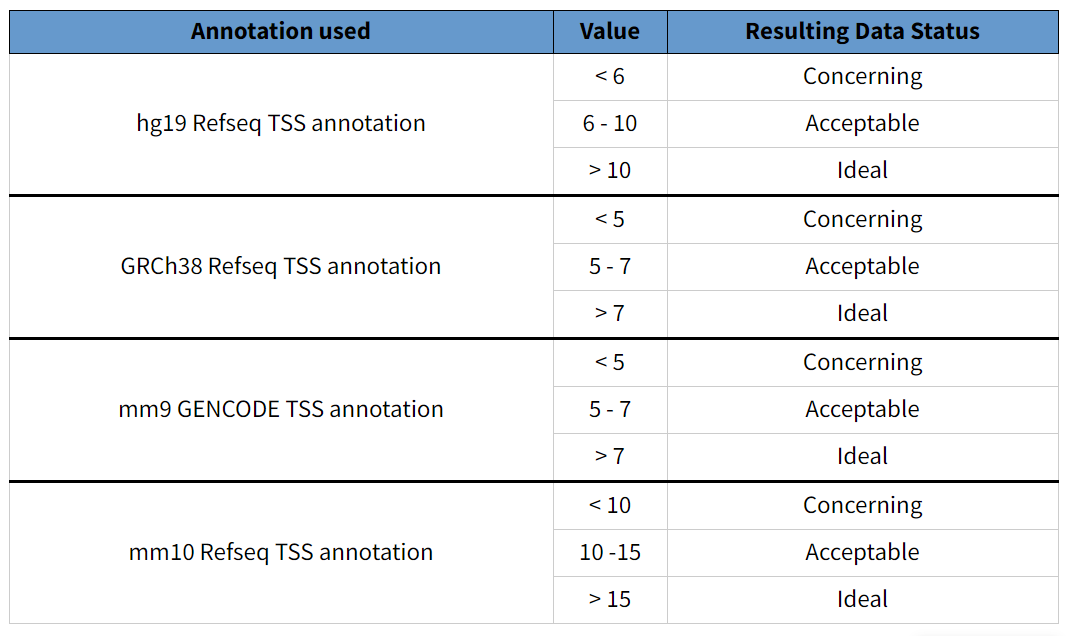
TSS标准化分数的计算方法不同,可能导致阈值偏差。10X的标准化方法是cut sites数除以最小值,而ENCODE的标准是在cut sites数除以两个末端各100bp的cut site数的平均值。由于一般越靠近中间数值越大,ENCODE的标准化分数整体比10x的分数小一些。
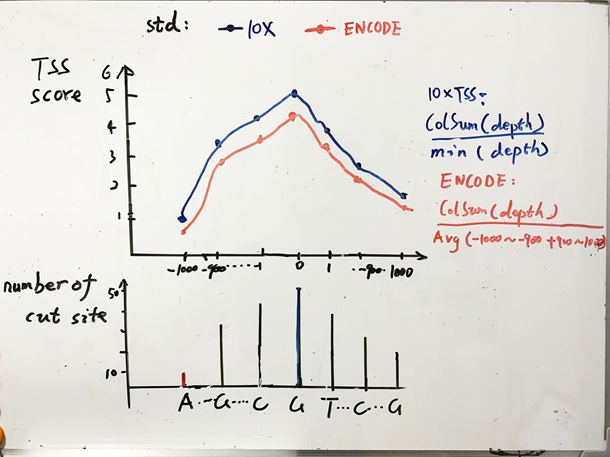
其他指标:Fraction of read pairs with a valid barcode > 75%;
- Q30 bases in R1 > 65%;
- Q30 bases in R2 > 65%;
- Q30 bases in barcode > 65%;
- Q30 bases in Sample Index > 90%;
- Estimated Number of Cells 500~10000 +- 20%;
- Median fragments per cell barcode > 500;
- Fragments in nucleosomefree regions >55%;
- Fraction of total read pairs mapped confidently to genome (>30 mapq) >80%;
- Fraction of total read pairs in mitochondria and in cell barcodes < 40%;
下游分析(以Signac为例)
Signac包由Seurat同一团队开发,独立于Seurat包,在2020年8月开始发布在GitHub上。目前仍是1.0.0版本[4]。
我们可以用10x官网的PBMC数据做演示。
首先加载所需R包:
library(Signac)library(Seurat)library(GenomeInfoDb)library(EnsDb.Hsapiens.v75)library(ggplot2)library(patchwork)set.seed(1234)
加载peaks, 细胞注释和片段分布数据,并创建object。这个object和Seurat object类似,只是在assay里多了peaks等信息。
counts <- Read10X_h5(filename = "atac_v1_pbmc_10k_filtered_peak_bc_matrix.h5")metadata <- read.csv(file = "atac_v1_pbmc_10k_singlecell.csv",header = TRUE,row.names = 1)chrom_assay <- CreateChromatinAssay(counts = counts,sep = c(":", "-"),genome = 'hg19',fragments = 'atac_v1_pbmc_10k_fragments.tsv.gz',min.cells = 10,min.features = 200)pbmc <- CreateSeuratObject(counts = chrom_assay,assay = "peaks",meta.data = metadata)
总共8728个细胞,87405个features。features不是基因,是基因组的注释区域,如启动子,增强子等。
pbmc[['peaks']]## ChromatinAssay data with 87405 features for 8728 cells## Variable features: 0## Genome: hg19## Annotation present: FALSE## Motifs present: FALSE## Fragment files: 1
加载注释:
# extract gene annotations from EnsDbannotations <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v75)# change to UCSC style since the data was mapped to hg19seqlevelsStyle(annotations) <- 'UCSC'genome(annotations) <- "hg19"# add the gene information to the objectAnnotation(pbmc) <- annotations
TSS和blacklist比例计算。
# compute nucleosome signal score per cellpbmc <- NucleosomeSignal(object = pbmc)# compute TSS enrichment score per cellpbmc <- TSSEnrichment(object = pbmc, fast = FALSE)# add blacklist ratio and fraction of reads in peakspbmc$pct_reads_in_peaks <- pbmc$peak_region_fragments / pbmc$passed_filters * 100pbmc$blacklist_ratio <- pbmc$blacklist_region_fragments / pbmc$peak_region_fragmentspbmc$high.tss <- ifelse(pbmc$TSS.enrichment > 2, 'High', 'Low')TSSPlot(pbmc, group.by = 'high.tss') + NoLegend()
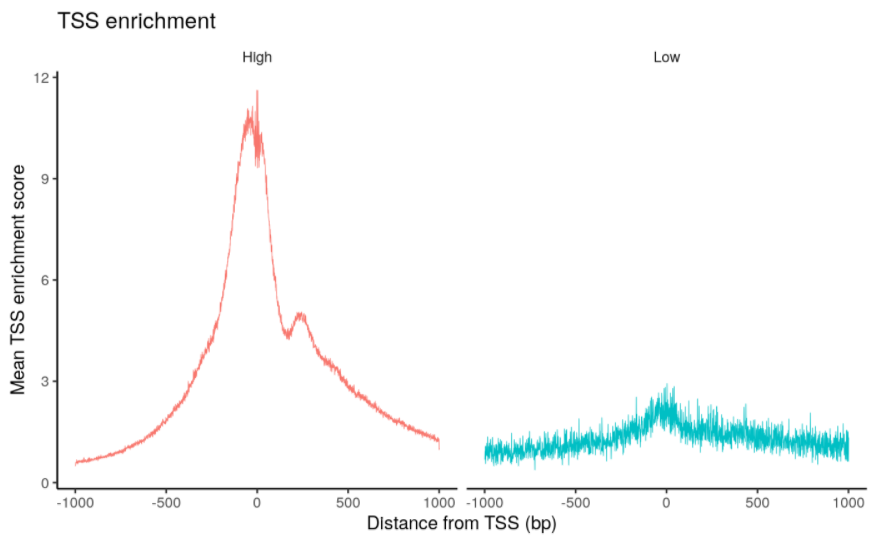
质控。Signac包用5个指标做过滤:
- TSS富集分数,大于2;
- blacklist比例,小于0.05;
- 缠绕核小体的片段与非核小体片段(< 147bp)的比例,小于4;
- 匹配peaks区域的片段比例,大于15%;
- 匹配peaks区域的片段数,大于3000,小于20000。
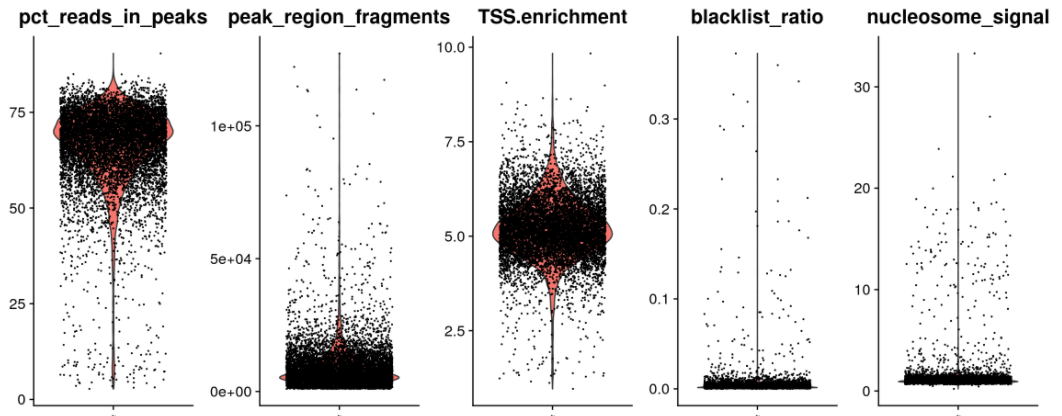
VlnPlot(object = pbmc,features = c('pct_reads_in_peaks', 'peak_region_fragments', 'TSS.enrichment', 'blacklist_ratio', 'nucleosome_signal'),pt.size = 0.1,ncol = 5)
 ```r
pbmc <- subset(
x = pbmc,
subset = peak_region_fragments > 3000 &
peak_region_fragments < 20000 &
pct_reads_in_peaks > 15 &
blacklist_ratio < 0.05 &
nucleosome_signal < 4 &
TSS.enrichment > 2
)
```r
pbmc <- subset(
x = pbmc,
subset = peak_region_fragments > 3000 &
peak_region_fragments < 20000 &
pct_reads_in_peaks > 15 &
blacklist_ratio < 0.05 &
nucleosome_signal < 4 &
TSS.enrichment > 2
)
pbmc
An object of class Seurat
87405 features across 7060 samples within 1 assay
Active assay: peaks (87405 features, 0 variable features)
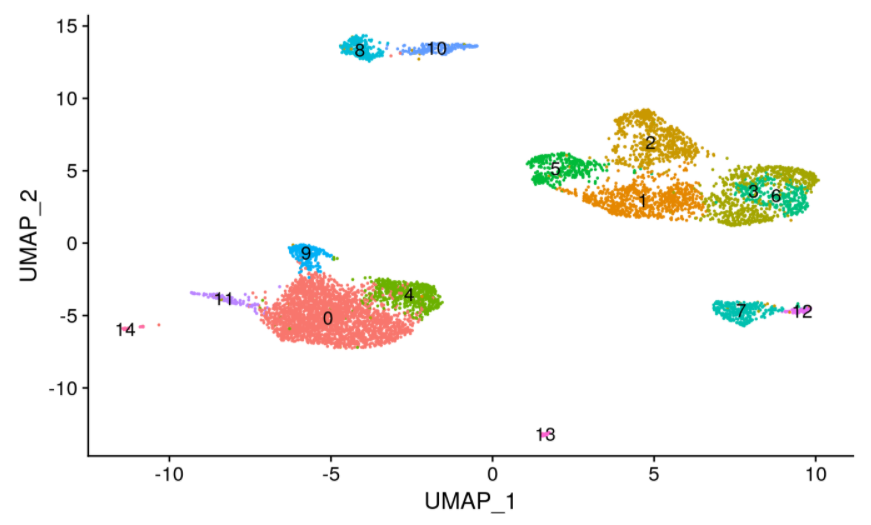
降维聚类。```rpbmc <- RunTFIDF(pbmc)pbmc <- FindTopFeatures(pbmc, min.cutoff = 'q0')pbmc <- RunSVD(pbmc)pbmc <- RunUMAP(object = pbmc, reduction = 'lsi', dims = 2:30)pbmc <- FindNeighbors(object = pbmc, reduction = 'lsi', dims = 2:30)pbmc <- FindClusters(object = pbmc, verbose = FALSE, algorithm = 3)DimPlot(object = pbmc, label = TRUE) + NoLegend()
创建基因活性矩阵。之前的聚类区域所用的features是peaks,为了展示不同分群基因活性的差异,首先要创建一个类似RNA表达的矩阵。用基因加上游2000bp区域的比对片段数代表该基因的活性。
gene.activities <- GeneActivity(pbmc)# add the gene activity matrix to the Seurat object as a new assay and normalize itpbmc[['RNA']] <- CreateAssayObject(counts = gene.activities)pbmc <- NormalizeData(object = pbmc,assay = 'RNA',normalization.method = 'LogNormalize',scale.factor = median(pbmc$nCount_RNA))
展示marker基因活性:
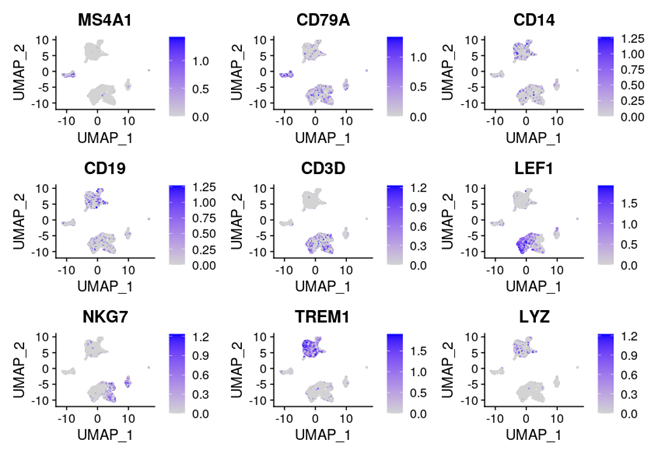
DefaultAssay(pbmc) <- 'RNA'FeaturePlot(object = pbmc,features = c('MS4A1', 'CD3D', 'LEF1', 'NKG7', 'TREM1', 'LYZ'),pt.size = 0.1,max.cutoff = 'q95',ncol = 3)

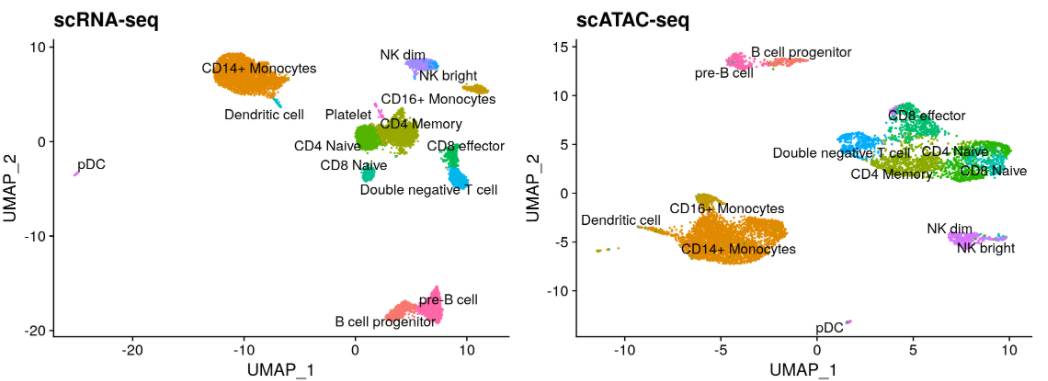
与scRNA-seq数据的整合分析。单细胞转录组数据地址:https://www.dropbox.com/s/zn6khirjafoyyxl/pbmc_10k_v3.rds?dl=0。
# Load the pre-processed scRNA-seq data for PBMCspbmc_rna <- readRDS("/home/stuartt/github/chrom/vignette_data/pbmc_10k_v3.rds")transfer.anchors <- FindTransferAnchors(reference = pbmc_rna,query = pbmc,reduction = 'cca')predicted.labels <- TransferData(anchorset = transfer.anchors,refdata = pbmc_rna$celltype,weight.reduction = pbmc[['lsi']],dims = 2:30)pbmc <- AddMetaData(object = pbmc, metadata = predicted.labels)

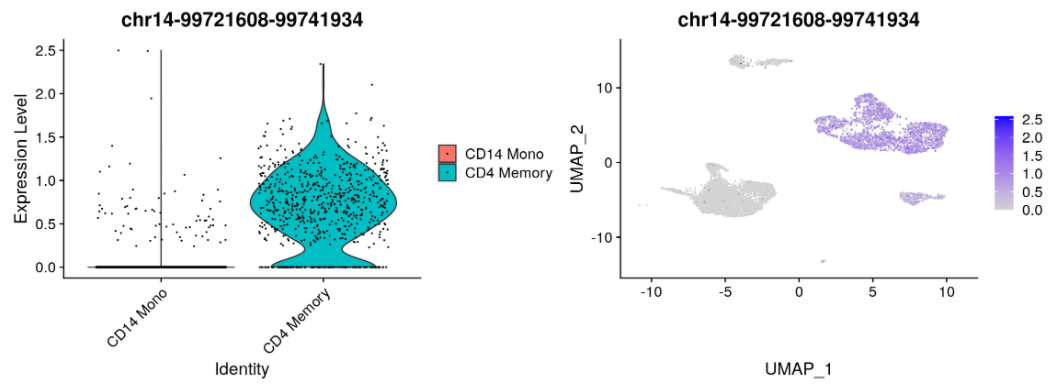
寻找细胞分群特异的peaks。
# change back to working with peaks instead of gene activitiesDefaultAssay(pbmc) <- 'peaks'da_peaks <- FindMarkers(object = pbmc,ident.1 = "CD4 Naive",ident.2 = "CD14 Mono",min.pct = 0.2,test.use = 'LR',latent.vars = 'peak_region_fragments')plot1 <- VlnPlot(object = pbmc,features = rownames(da_peaks)[1],pt.size = 0.1,idents = c("CD4 Memory","CD14 Mono"))plot2 <- FeaturePlot(object = pbmc,features = rownames(da_peaks)[1],pt.size = 0.1)plot1 | plot2

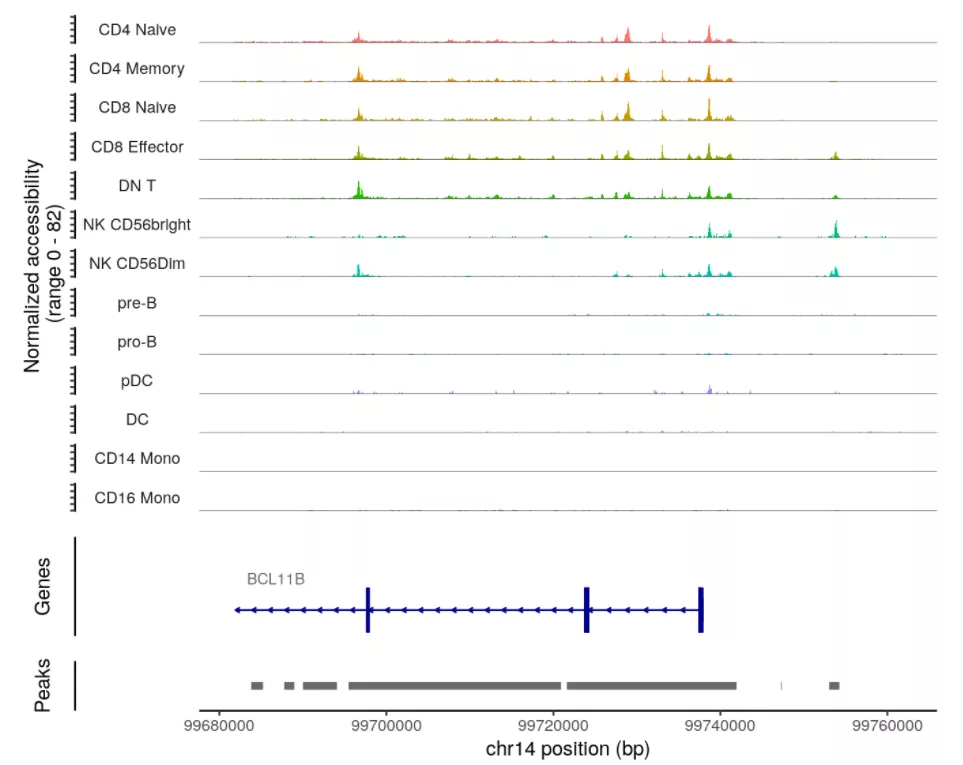
展示基因在不同细胞类型的开放程度:
# set plotting orderlevels(pbmc) <- c("CD4 Naive","CD4 Memory","CD8 Naive","CD8 Effector","DN T","NK CD56bright","NK CD56Dim","pre-B",'pro-B',"pDC","DC","CD14 Mono",'CD16 Mono')CoveragePlot(object = pbmc,region = rownames(da_peaks)[1],extend.upstream = 40000,extend.downstream = 20000)
此外还有其他分析,如TF footprinting等。footprinting顾名思义是指转录因子留下的印记,由于Tn5酶不能剪切到TF结合的区域,所以footprinting图相对与TSS图,中间有“凹陷”,凹陷的程度根据TF结合的时间确定[6]。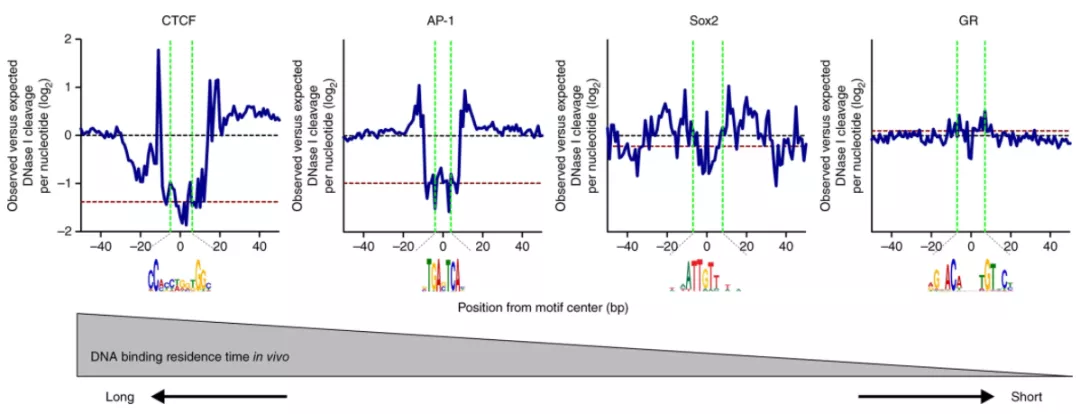
总的来说,Signac包是一个亲民实用的工具。虽然有一些不足的地方,如染色体的注释目前只能选择UCSC的,不能选Ensemble。
参考文献:
[1] Buenrostro, J., Giresi, P., Zaba, L. et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218 (2013). https://doi.org/10.1038/nmeth.2688
[2] Buenrostro, J., Wu, B., Litzenburger, U. et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). https://doi.org/10.1038/nature14590
[3] https://www.encodeproject.org/atac-seq/
[4] https://satijalab.org/signac/index.html
[5]https://assets.ctfassets.net/an68im79xiti/Cts31zFxXFXVwJ1lzU3Pc/fe66343ffd3039de73ecee6a1a6f5b7b/CG000202_TechnicalNote_InterpretingCellRangerATACWebSummaryFiles_Rev-A.pdf
[6] Sung, M., Baek, S. & Hager, G. Genome-wide footprinting: ready for prime time?. Nat Methods 13, 222–228 (2016). https://doi.org/10.1038/nmeth.3766

若有收获,就点个赞吧
0 人点赞
