- 往期精选
- Running Cicero 运行Cicero
- Visualizing Cicero Connections 可视化Cicero得到的连接
- Comparing Cicero connections to other datasets 比较Cicero连接和其他数据集
- Finding cis-Co-accessibility Networks (CCANS) 构建顺式共可及性网络
- Cicero gene activity scores 计算Cicero基因活性得分
- Advanced visualizaton 高级可视化技巧
- Important Considerations for Non-Human Data

往期精选
使用Signac包进行单细胞ATAC-seq数据分析(一):Analyzing PBMC scATAC-seq
使用Signac包进行单细胞ATAC-seq数据分析(二):Motif analysis with Signac
使用Signac包进行单细胞ATAC-seq数据分析(三):scATAC-seq data integration
使用Signac包进行单细胞ATAC-seq数据分析(四):Merging objects
使用Cicero包进行单细胞ATAC-seq分析(一):Cicero introduction and installing
在本教程中,我们将学习使用Cicero基于单细胞ATAC-seq数据构建顺式调控元件互作网络。
Running Cicero 运行Cicero
Create a Cicero CDS
由于单细胞染色质可及性的数据通常非常稀疏,因此我们需要聚合相似的细胞以创建更密集的计数数据,来准确的计算评估共可及性(co-accessibility)分数。Cicero使用k最近邻方法来创建一个重叠的细胞集,并基于细胞相似度的降维坐标图(如来自tSNE或DDRTree图)来构建这些集合。
这里,我们将以tSNE和DDRTree降维方法进行展示,这两种降维方法均可从Monocle(由Cicero加载)获得。数据降维后,我们可以使用make_cicero_cds函数创建聚合的CDS对象。make_cicero_cds函数以CDS对象和降维后的坐标为输入,降维后的坐标reduce_coordinates应该采用data.frame或矩阵的形式,其中行名称与CDS的pData表中的细胞ID相匹配,列为降维对象的坐标,例如: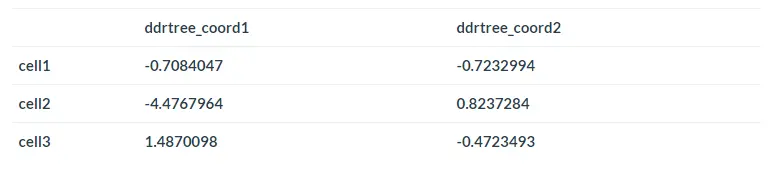
set.seed(2017)input_cds <- detectGenes(input_cds)input_cds <- estimateSizeFactors(input_cds)# 使用tSNE方法进行数据降维input_cds <- reduceDimension(input_cds, max_components = 2, num_dim=6,reduction_method = 'tSNE', norm_method = "none")# 提取tSNE降维后的坐标tsne_coords <- t(reducedDimA(input_cds))row.names(tsne_coords) <- row.names(pData(input_cds))# 使用make_cicero_cds函数构建cicero CDS对象cicero_cds <- make_cicero_cds(input_cds, reduced_coordinates = tsne_coords)
Run Cicero
Cicero包的主要功能是通过计算评估全基因组中位点之间的共可及性,从而预测顺式调控相互作用。
Cicero提供了两种方法来获取此信息:
run_cicero:get Cicero outputs with all defaults(使用所有默认值获得Cicero的输出)。run_cicero函数将使用默认值调用Cicero代码的每个相关部分,并在计算过程中找出最佳估计参数。对于大多数用户来说,这将是最好的起点。- Call functions separately, for more flexibility(分别调用函数,以提高灵活性)。对于希望在调用的参数上具有更大的灵活性,以及想要访问中间信息的用户,Cicero允许我们分别调用每个组件。

(一)直接调用run_cicero函数(Recommended)
使用Cicero计算获得共可及性得分的最简单方法是直接调用run_cicero函数。该函数需要一个cicero CDS对象和一个基因组坐标文件(里面包含所用参考基因组的每个染色体的长度)作为输入。Cicero包中提供了人类hg19的坐标,可以通过data(“human.hg19.genome”)获取访问。
# 加载人hg19参考基因组的坐标信息data("human.hg19.genome")# 提取18号染色体的信息sample_genome <- subset(human.hg19.genome, V1 == "chr18")# 直接使用run_cicero函数计算共可及性得分conns <- run_cicero(cicero_cds, sample_genome) # Takes a few minutes to run# 查看运行的结果head(conns)Peak1 Peak2 coaccesschr18_10025_10225 chr18_104385_104585 0.03791526chr18_10025_10225 chr18_10603_11103 0.86971957chr18_10025_10225 chr18_111867_112367 0.00000000
(二)分别调用Cicero的不同函数(Alternative)
我们还可以使用一种替代的方法分别调用Cicero流程各个部分的函数,构成Cicero流程的主要有三个功能:
estimate_distance_parameter. 该函数基于基因组的随机小窗口计算距离惩罚参数。generate_cicero_models. 此函数使用上一步确定的距离参数,并使用图形化LASSO模型基于距离的惩罚来计算基因组重叠窗口的共可及性得分。assemble_connections. 此函数将generate_cicero_models的输出作为输入,并协调重叠的模型以创建最终共可及性得分的列表。
以下是每个函数相关参数的解释说明: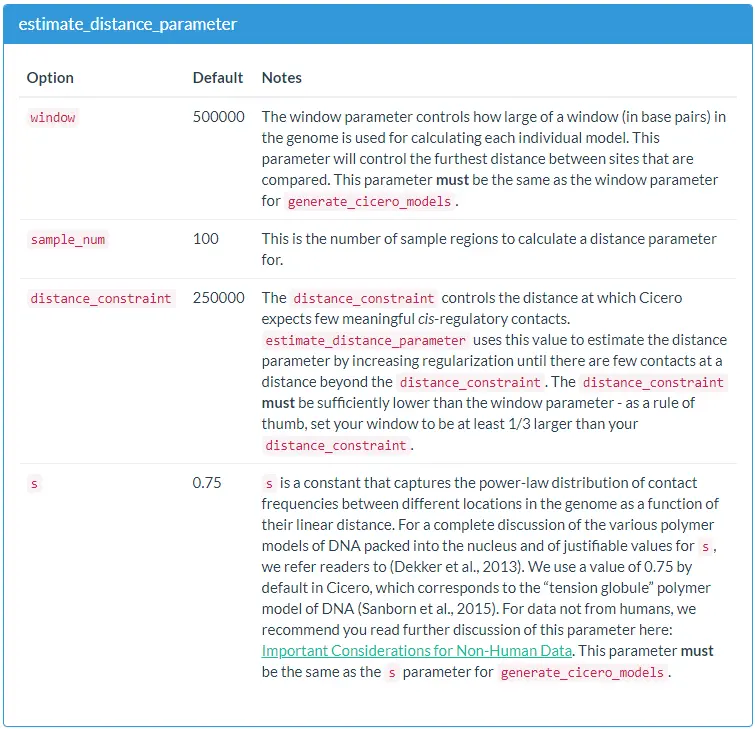
Visualizing Cicero Connections 可视化Cicero得到的连接
Cicero包提供了一个通用的绘图功能,可以使用plot_connections函数可视化共可及性。此函数使用Gviz包的框架绘制基因组浏览器样式的图。我们改编了Sushi R包中的函数来映射连接。plot_connections函数有很多参数,其中一些在“高级可视化”部分中进行了详细介绍。
# 加载基因注释文件data(gene_annotation_sample)# 使用plot_connections进行可视化plot_connections(conns, "chr18", 8575097, 8839855,gene_model = gene_annotation_sample,coaccess_cutoff = .25,connection_width = .5,collapseTranscripts = "longest" )
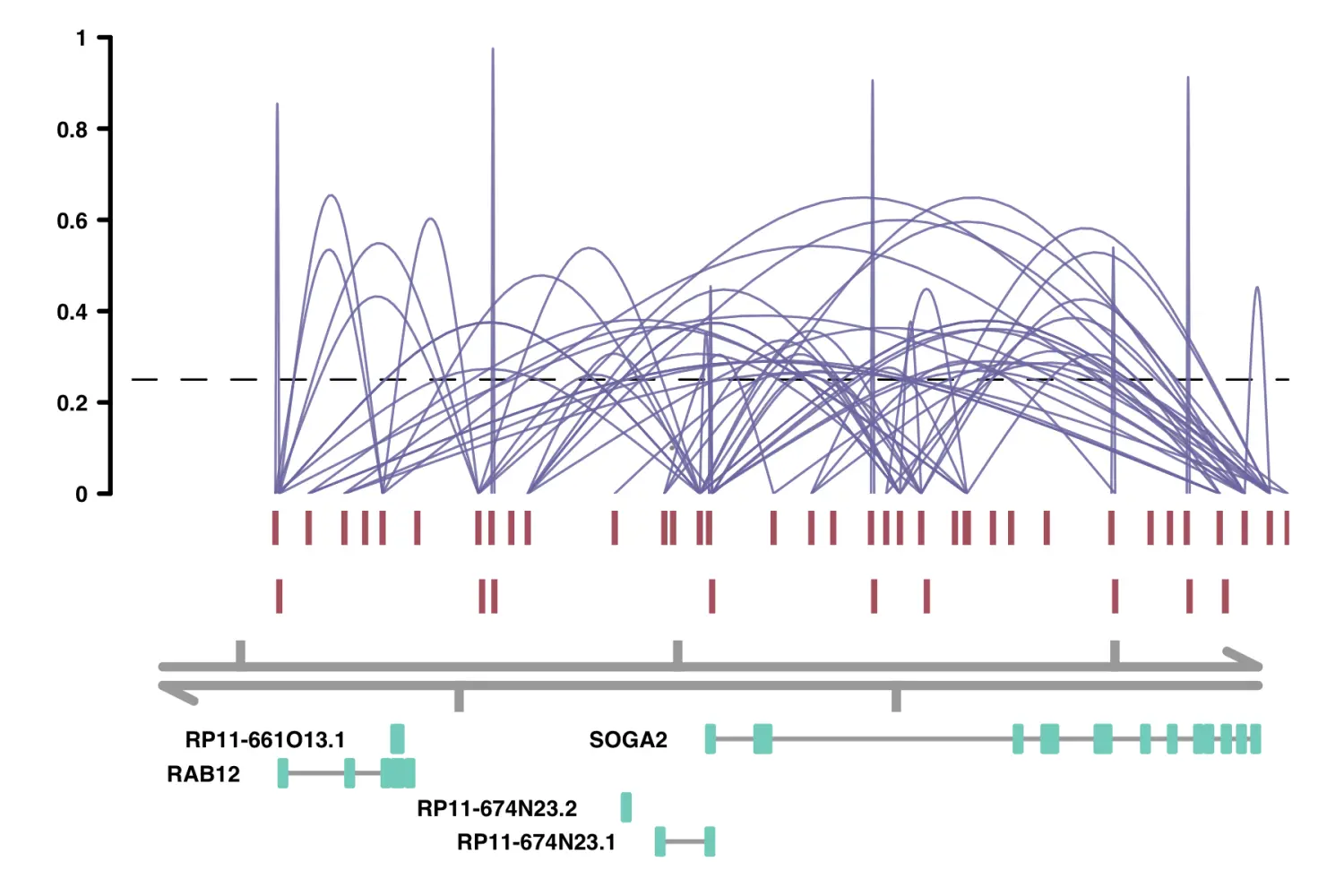
Comparing Cicero connections to other datasets 比较Cicero连接和其他数据集
通常,将Cicero计算得到的连接(connections)与其他具有类似连接类型的数据集进行比较非常有用。例如,我们可能想将Cicero的输出与ChIA-PET的连接进行比较。为此,Cicero提供了一个名为compare_connections的函数,此函数以两个数据框的连接对(connection pairs)conns1和conns2作为输入,并从conns2中找到的conns1返回连接的逻辑向量。它使用GenomicRanges包进行此功能的比较,并使用该程序包中的max_gap参数允许比较时出现斜率。
例如,如果我们想将Cicero预测的连接结果与一组(虚构的)ChIA-PET连接数据进行比较,则可以运行:
# 构建ChIA-PET的连接数据chia_conns <- data.frame(Peak1 = c("chr18_10000_10200", "chr18_10000_10200","chr18_49500_49600"),Peak2 = c("chr18_10600_10700", "chr18_111700_111800","chr18_10600_10700"))# 查看连接数据head(chia_conns)# Peak1 Peak2# 1 chr18_10000_10200 chr18_10600_10700# 2 chr18_10000_10200 chr18_111700_111800# 3 chr18_49500_49600 chr18_10600_10700# 使用compare_connections函数进行比较conns$in_chia <- compare_connections(conns, chia_conns)# 查看比较后的结果head(conns)# Peak1 Peak2 coaccess in_chia# 2 chr18_10025_10225 chr18_10603_11103 0.85209126 TRUE# 3 chr18_10025_10225 chr18_11604_13986 -0.55433268 FALSE# 4 chr18_10025_10225 chr18_49557_50057 -0.43594546 FALSE# 5 chr18_10025_10225 chr18_50240_50740 -0.43662436 FALSE# 6 chr18_10025_10225 chr18_104385_104585 0.00000000 FALSE# 7 chr18_10025_10225 chr18_111867_112367 0.01405174 FALSE
当比较两个完全不同的数据集时,我们可能会发现这种重叠过于严格。仔细观察,ChIA-PET数据的第二行与conns的最后一行非常接近,差异仅约67个碱基对,这可能是peak-calling的问题,我们可以调整max_gap参数:
conns$in_chia_100 <- compare_connections(conns, chia_conns, maxgap=100)head(conns)# Peak1 Peak2 coaccess in_chia in_chia_100# 2 chr18_10025_10225 chr18_10603_11103 0.85209126 TRUE TRUE# 3 chr18_10025_10225 chr18_11604_13986 -0.55433268 FALSE FALSE# 4 chr18_10025_10225 chr18_49557_50057 -0.43594546 FALSE FALSE# 5 chr18_10025_10225 chr18_50240_50740 -0.43662436 FALSE FALSE# 6 chr18_10025_10225 chr18_104385_104585 0.00000000 FALSE FALSE# 7 chr18_10025_10225 chr18_111867_112367 0.01405174 FALSE TRUE
此外,Cicero的绘图功能还可以直观地比较不同的数据集。为此,请使用compare_track参数。用来比较的连接数据必须包括前两个峰值列之外的第三列,称为“coaccess”,该值决定了绘图中连接的高度。这可能是一种定量方法,例如ChIA-PET中的连接数。
# Add a column of 1s called "coaccess"# 构建ChIA-PET连接数据chia_conns <- data.frame(Peak1 = c("chr18_10000_10200", "chr18_10000_10200","chr18_49500_49600"),Peak2 = c("chr18_10600_10700", "chr18_111700_111800","chr18_10600_10700"),coaccess = c(1, 1, 1))# 使用plot_connections函数进行可视化plot_connections(conns, "chr18", 10000, 112367,gene_model = gene_annotation_sample,coaccess_cutoff = 0,connection_width = .5,comparison_track = chia_conns,include_axis_track = F,collapseTranscripts = "longest")
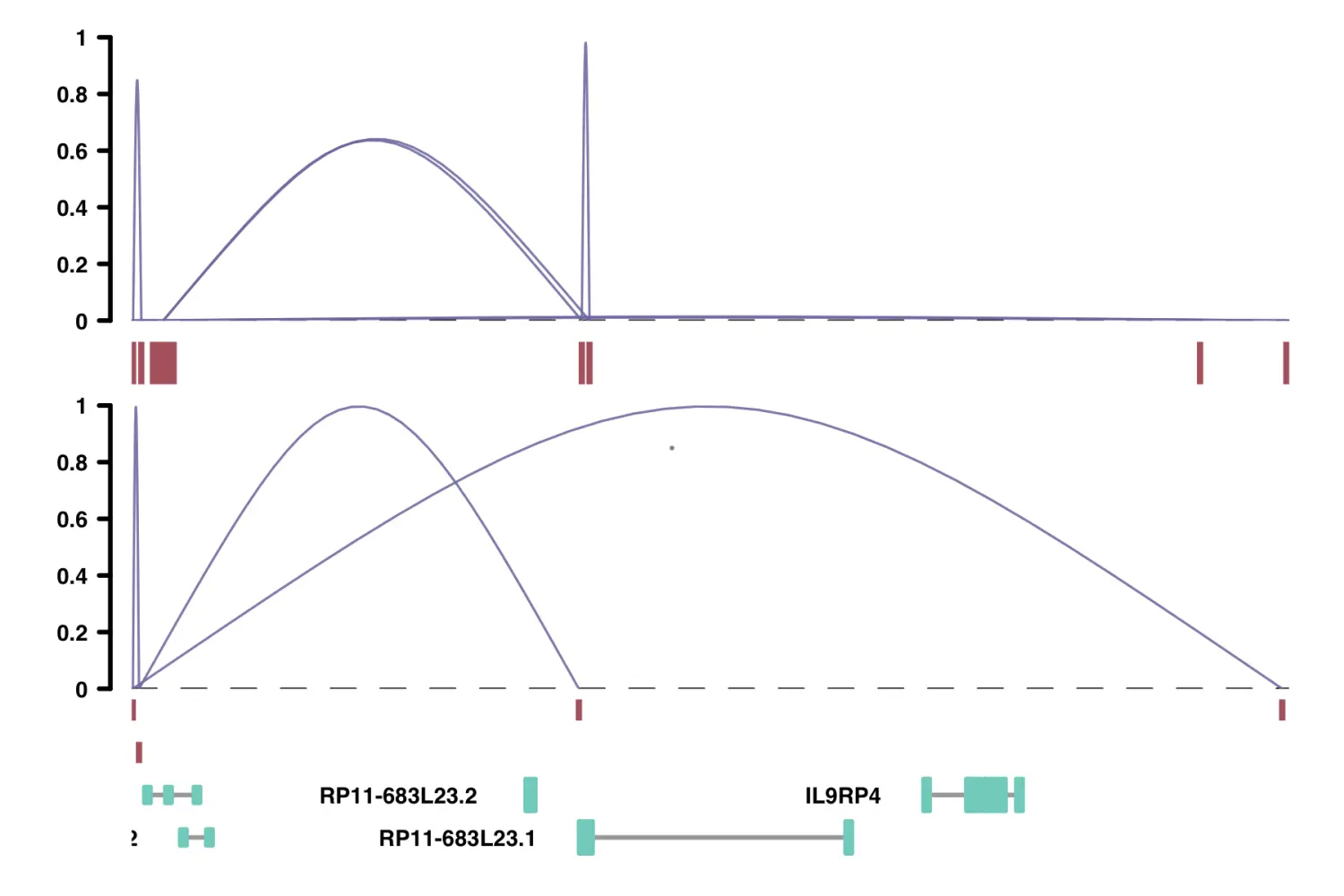
Finding cis-Co-accessibility Networks (CCANS) 构建顺式共可及性网络
除了计算成对的共可及性得分,Cicero还可以用来构建顺式共可及性网络Cis-Co-accessibility Networks(CCAN),CCAN是彼此高度共可及性互作模块。我们使用Louvain检测算法(Blondel等人,2008)来查找倾向于共可及性的互作集群。函数generate_ccans将连接数据作为输入,并为每个输入峰值输出CCAN评分。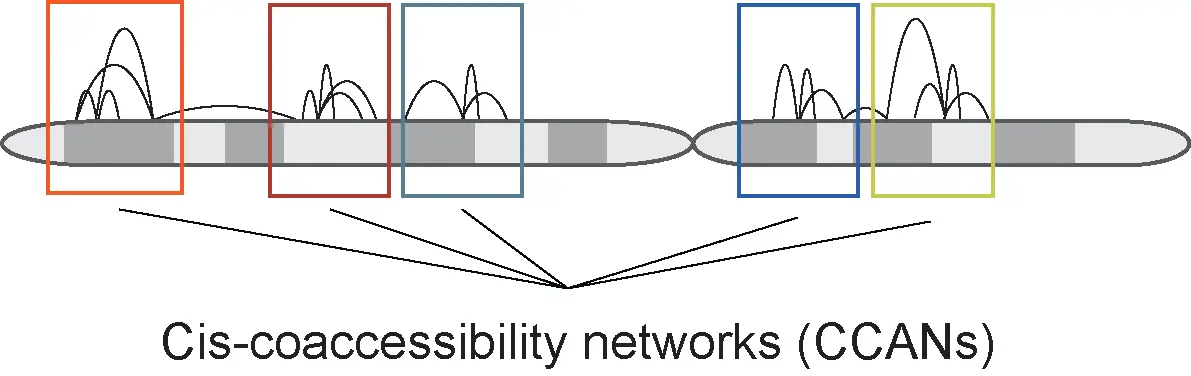
generate_ccans函数有一个coaccess_cutoff_override参数,当该参数设置为NULL时,该函数将根据不同cutoff的总CCAN数量,确定并报告CCAN生成的合适的共可及性得分的阈值。我们还可以将coaccess_cutoff_override值设置为介于0和1之间的数字,以覆盖函数的cutoff-find部分。如果您觉得自动找到的临界值太严格或太松散,或者如果您正在重新运行代码并知道临界值,则此选项很有用,因为临界值的查找过程可能会很慢。
# 使用generate_ccans函数构建CCANCCAN_assigns <- generate_ccans(conns)# [1] "Co-accessibility cutoff used: 0.24"# 查看计算的结果head(CCAN_assigns)# Peak CCAN# chr18_10025_10225 chr18_10025_10225 1# chr18_10603_11103 chr18_10603_11103 1# chr18_11604_13986 chr18_11604_13986 1# chr18_49557_50057 chr18_49557_50057 1# chr18_50240_50740 chr18_50240_50740 1# chr18_157883_158536 chr18_157883_158536 1
Cicero gene activity scores 计算Cicero基因活性得分
我们发现,直接通过启动子的可及性预测基因的表达,其结果往往不是很好。但是,使用Cicero得到的连接,我们可以更好地了解启动子及其相关远端位置的总体可及性。这些区域可及性的综合得分与基因表达具有更好的一致性。我们称此分数为Cicero基因活性分数,它是使用两个函数计算得出的。
初始函数称为build_gene_activity_matrix。此函数以CDS对象和Cicero连接列表为输入,计算得出未标准化的基因活性得分表格。注意,在输入的CDS对象中必须在fData表中有一列称为“基因”,如果该peak是启动子,则指示该基因;如果该peak是在远端,则指示为NA。
使用build_gene_activity_matrix函数计算得到的基因活性得分是未标准化的,我们还需使用第二个函数normalize_gene_activities对其进行标准化处理。如果要比较不同数据子集中的基因活性,则应通过传入未归一化的矩阵列表来对所有基因活性子集进行归一化。如果只希望对一个矩阵进行归一化,只需将其自己传递给函数。标准化后的基因活性得分范围是0到1。
#### Add a column for the fData table indicating the gene if a peak is a promoter ##### Create a gene annotation set that only marks the transcription start sites of the genes. We use this as a proxy for promoters.# To do this we need the first exon of each transcriptpos <- subset(gene_annotation_sample, strand == "+")pos <- pos[order(pos$start),]pos <- pos[!duplicated(pos$transcript),] # remove all but the first exons per transcriptpos$end <- pos$start + 1 # make a 1 base pair marker of the TSSneg <- subset(gene_annotation_sample, strand == "-")neg <- neg[order(neg$start, decreasing = TRUE),]neg <- neg[!duplicated(neg$transcript),] # remove all but the first exons per transcriptneg$start <- neg$end - 1gene_annotation_sub <- rbind(pos, neg)# Make a subset of the TSS annotation columns containing just the coordinates and the gene namegene_annotation_sub <- gene_annotation_sub[,c(1:3, 8)]# Rename the gene symbol column to "gene"names(gene_annotation_sub)[4] <- "gene"input_cds <- annotate_cds_by_site(input_cds, gene_annotation_sub)head(fData(input_cds))# site_name chr bp1 bp2 num_cells_expressed overlap gene# chr18_10025_10225 chr18_10025_10225 18 10025 10225 5 NA <NA># chr18_10603_11103 chr18_10603_11103 18 10603 11103 1 1 AP005530.1# chr18_11604_13986 chr18_11604_13986 18 11604 13986 9 203 AP005530.1# chr18_49557_50057 chr18_49557_50057 18 49557 50057 2 331 RP11-683L23.1# chr18_50240_50740 chr18_50240_50740 18 50240 50740 2 129 RP11-683L23.1# chr18_104385_104585 chr18_104385_104585 18 104385 104585 1 NA <NA>#### Generate gene activity scores ##### generate unnormalized gene activity matrixunnorm_ga <- build_gene_activity_matrix(input_cds, conns)# remove any rows/columns with all zeroesunnorm_ga <- unnorm_ga[!Matrix::rowSums(unnorm_ga) == 0, !Matrix::colSums(unnorm_ga) == 0]# make a list of num_genes_expressednum_genes <- pData(input_cds)$num_genes_expressednames(num_genes) <- row.names(pData(input_cds))# normalizecicero_gene_activities <- normalize_gene_activities(unnorm_ga, num_genes)# if you had two datasets to normalize, you would pass both:# num_genes should then include all cells from both setsunnorm_ga2 <- unnorm_gacicero_gene_activities <- normalize_gene_activities(list(unnorm_ga, unnorm_ga2), num_genes)
Advanced visualizaton 高级可视化技巧
Some useful parameters
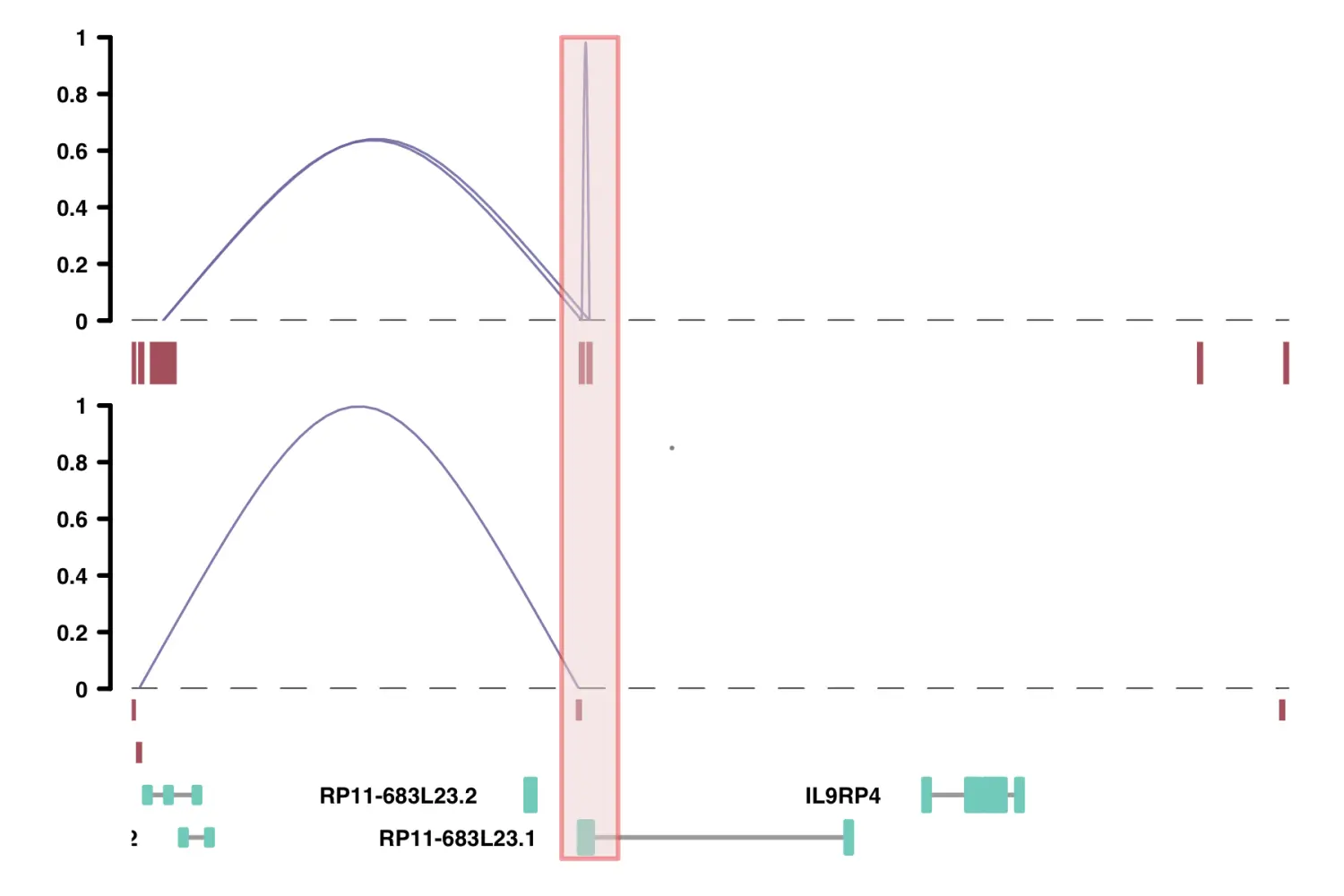
Viewpoints:Viewpoints参数可以让我们仅查看来自基因组中特定位置的连接情况。这在与4C-seq数据进行比较时可能很有用。
# viewpoint = "chr18_48000_53000"参数指定可视化特定区域plot_connections(conns, "chr18", 10000, 112367,viewpoint = "chr18_48000_53000",gene_model = gene_annotation_sample,coaccess_cutoff = 0,connection_width = .5,comparison_track = chia_conns,include_axis_track = F,collapseTranscripts = "longest")
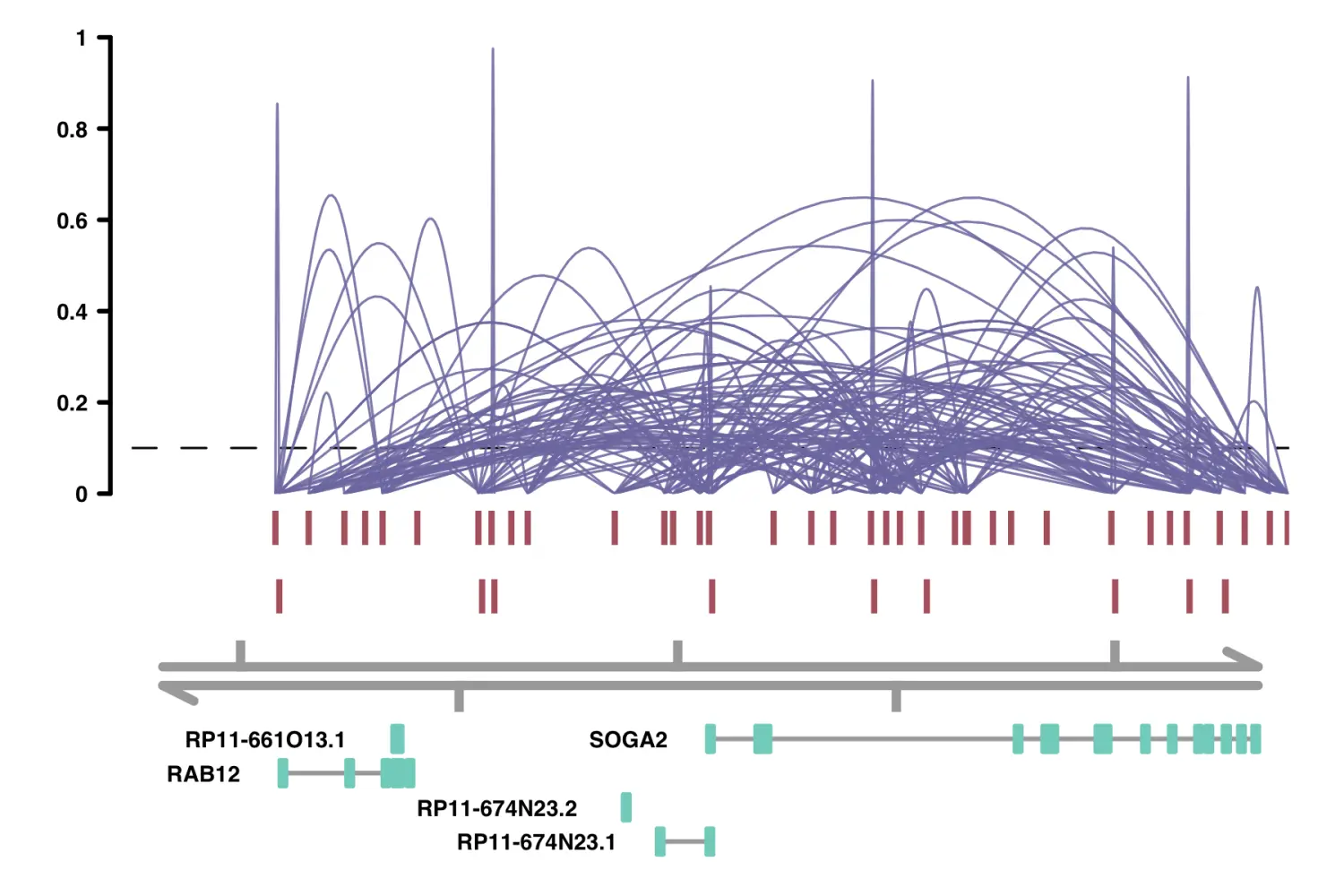
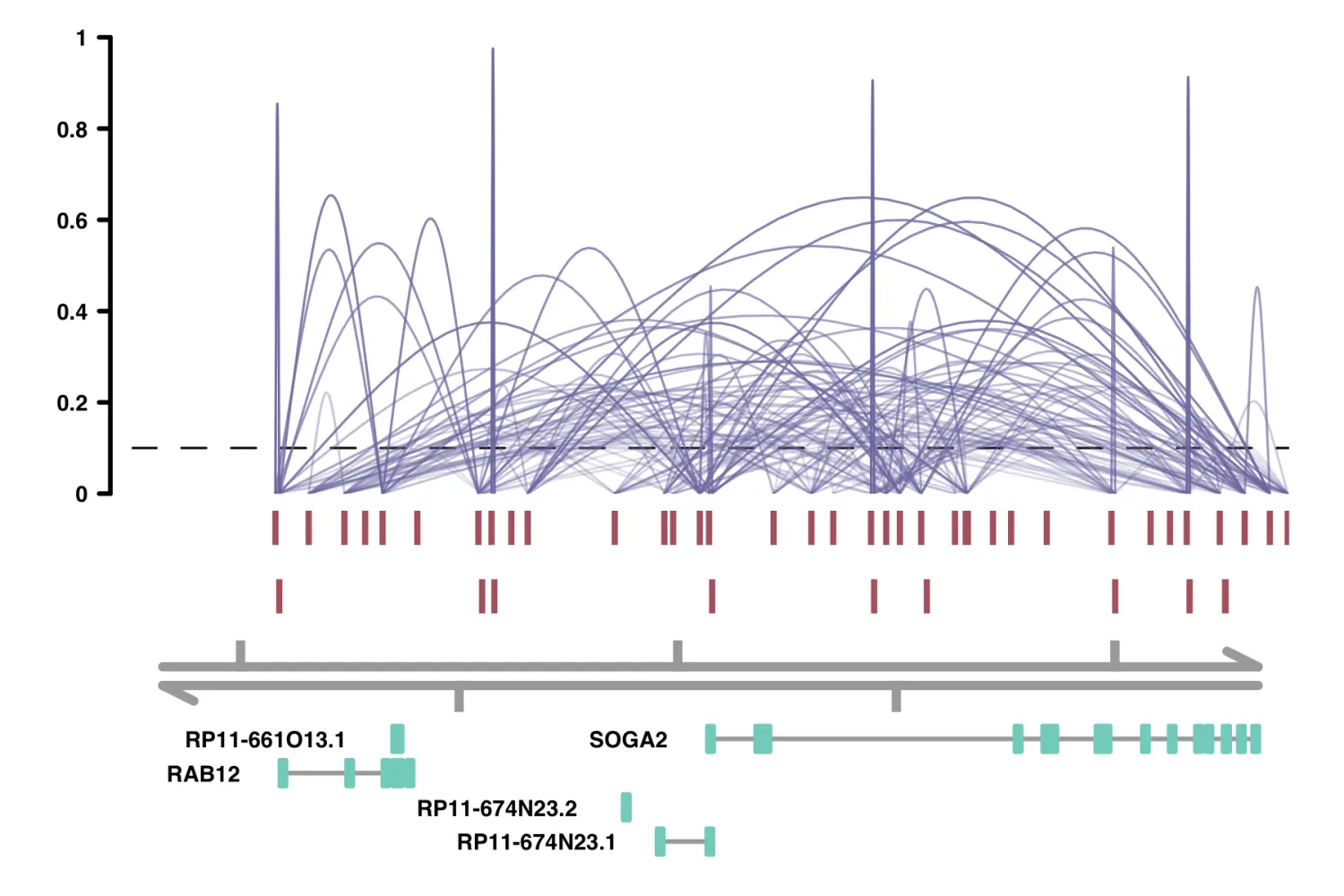
alpha_by_coaccess: alpha_by_coaccess参数可以使连接曲线的Alpha(透明度)基于共可及性的大小进行缩放。比较以下两个图。
# alpha_by_coaccess = FALSEplot_connections(conns,alpha_by_coaccess = FALSE,"chr18", 8575097, 8839855,gene_model = gene_annotation_sample,coaccess_cutoff = 0.1,connection_width = .5,collapseTranscripts = "longest" )# alpha_by_coaccess = TRUEplot_connections(conns,alpha_by_coaccess = TRUE,"chr18", 8575097, 8839855,gene_model = gene_annotation_sample,coaccess_cutoff = 0.1,connection_width = .5,collapseTranscripts = "longest" )
Colors: plot_connections函数提供了许多用于设定不同类型颜色的参数。
peak_color
comparison_peak_color
connection_color
comparison_connection_color
gene_model_color
viewpoint_color
viewpoint_fill
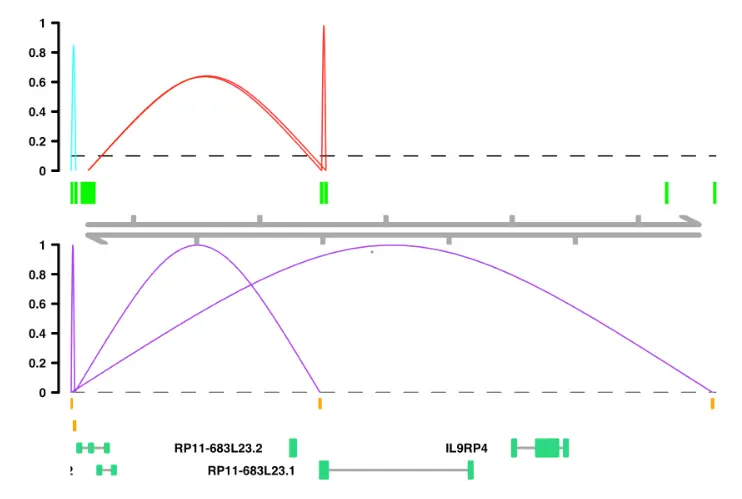
可以使用颜色的名称(如:“blue”)或颜色代码(如:“#B4656F”)的形式为每个参数提供颜色值。 另外,对于前四个参数,我们可以使用conns文件中提供颜色值的列。
# When the color column is not already colors, random colors are assignedplot_connections(conns,"chr18", 10000, 112367,connection_color = "in_chia_100",comparison_track = chia_conns,peak_color = "green",comparison_peak_color = "orange",comparison_connection_color = "purple",gene_model_color = "#2DD881",gene_model = gene_annotation_sample,coaccess_cutoff = 0.1,connection_width = .5,collapseTranscripts = "longest" )
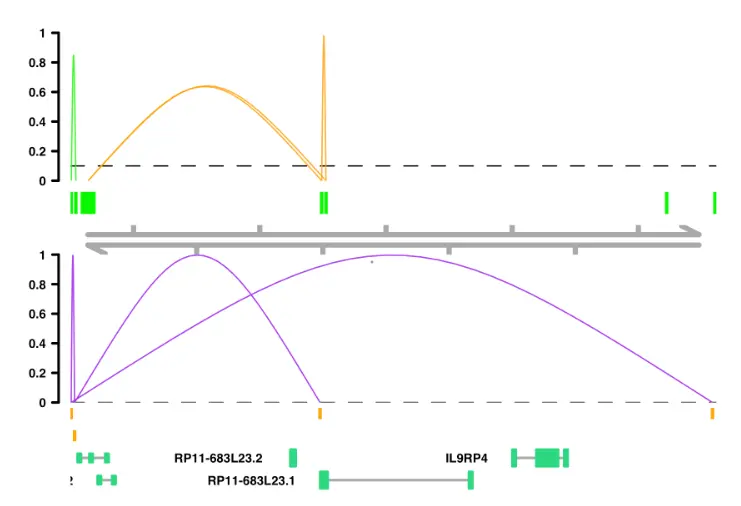
# If I want specific color scheme, I can make a column of color namesconns$conn_color <- "orange"conns$conn_color[conns$in_chia_100] <- "green"plot_connections(conns,"chr18", 10000, 112367,connection_color = "conn_color",comparison_track = chia_conns,peak_color = "green",comparison_peak_color = "orange",comparison_connection_color = "purple",gene_model_color = "#2DD881",gene_model = gene_annotation_sample,coaccess_cutoff = 0.1,connection_width = .5,collapseTranscripts = "longest" )
# For coloring Peaks, I need the color column to correspond to Peak1:conns$peak1_color <- FALSEconns$peak1_color[conns$Peak1 == "chr18_11604_13986"] <- TRUEplot_connections(conns,"chr18", 10000, 112367,connection_color = "green",comparison_track = chia_conns,peak_color = "peak1_color",comparison_peak_color = "orange",comparison_connection_color = "purple",gene_model_color = "#2DD881",gene_model = gene_annotation_sample,coaccess_cutoff = 0.1,connection_width = .5,collapseTranscripts = "longest" )
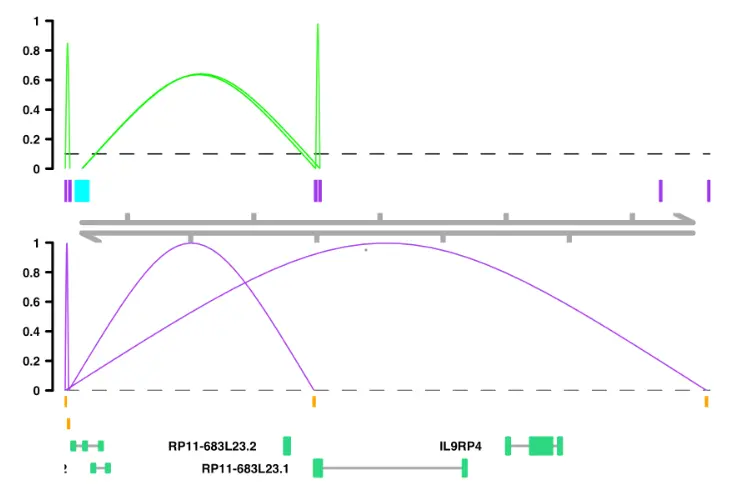
- Customizing everything with
return_as_list
如果设置return_as_list参数为TRUE,将不会绘制出图像,而是返回一个图像列表。
# 设置return_as_list = TRUEplot_list <- plot_connections(conns,"chr18", 10000, 112367,gene_model = gene_annotation_sample,coaccess_cutoff = 0.1,connection_width = .5,collapseTranscripts = "longest",return_as_list = TRUE)plot_list# [[1]]# CustomTrack ''# [[2]]# AnnotationTrack 'Peaks'# | genome: NA# | active chromosome: chr18# | annotation features: 7# [[3]]# Genome axis 'Axis'# [[4]]# GeneRegionTrack 'Gene Model'# | genome: NA# | active chromosome: chr18# | annotation features: 33
这将按绘图顺序返回一个由各个图形元件组成的列表。首先是CustomTrack,它是Cicero连接图形,其次是峰注释轨道(peak annotation track),再就是基因组轴轨道(genome axis track),最后是基因模型轨道(gene model track)。现在,我可以使用Gviz包添加其他的track,并重新排列tracks或替换tracks:
conservation <- UcscTrack(genome = "hg19", chromosome = "chr18",track = "Conservation", table = "phyloP100wayAll",fontsize.group=6,fontsize=6, cex.axis=.8,from = 10000, to = 112367, trackType = "DataTrack",start = "start", end = "end", data = "score", size = .1,type = "histogram", window = "auto", col.histogram = "#587B7F",fill.histogram = "#587B7F", ylim = c(-1, 2.5),name = "Conservation")# I will replace the genome axis track with a track on conservation valuesplot_list[[3]] <- conservation# To make the plot, I will now use Gviz's plotTracks function# The included options are the defaults in plot_connections,# but all can be modified according to Gviz's documentation# The main new paramter that you must include, is the sizes# parameter. This parameter controls what proportion of the# height of your plot is allocated for each track. The sizes# parameter must be a vector of the same length as plot_listGviz::plotTracks(plot_list,sizes = c(2,1,1,1),from = 10000, to = 112367, chromosome = "chr18",transcriptAnnotation = "symbol",col.axis = "black",fontsize.group = 6,fontcolor.legend = "black",lwd=.3,title.width = .5,background.title = "transparent",col.border.title = "transparent")
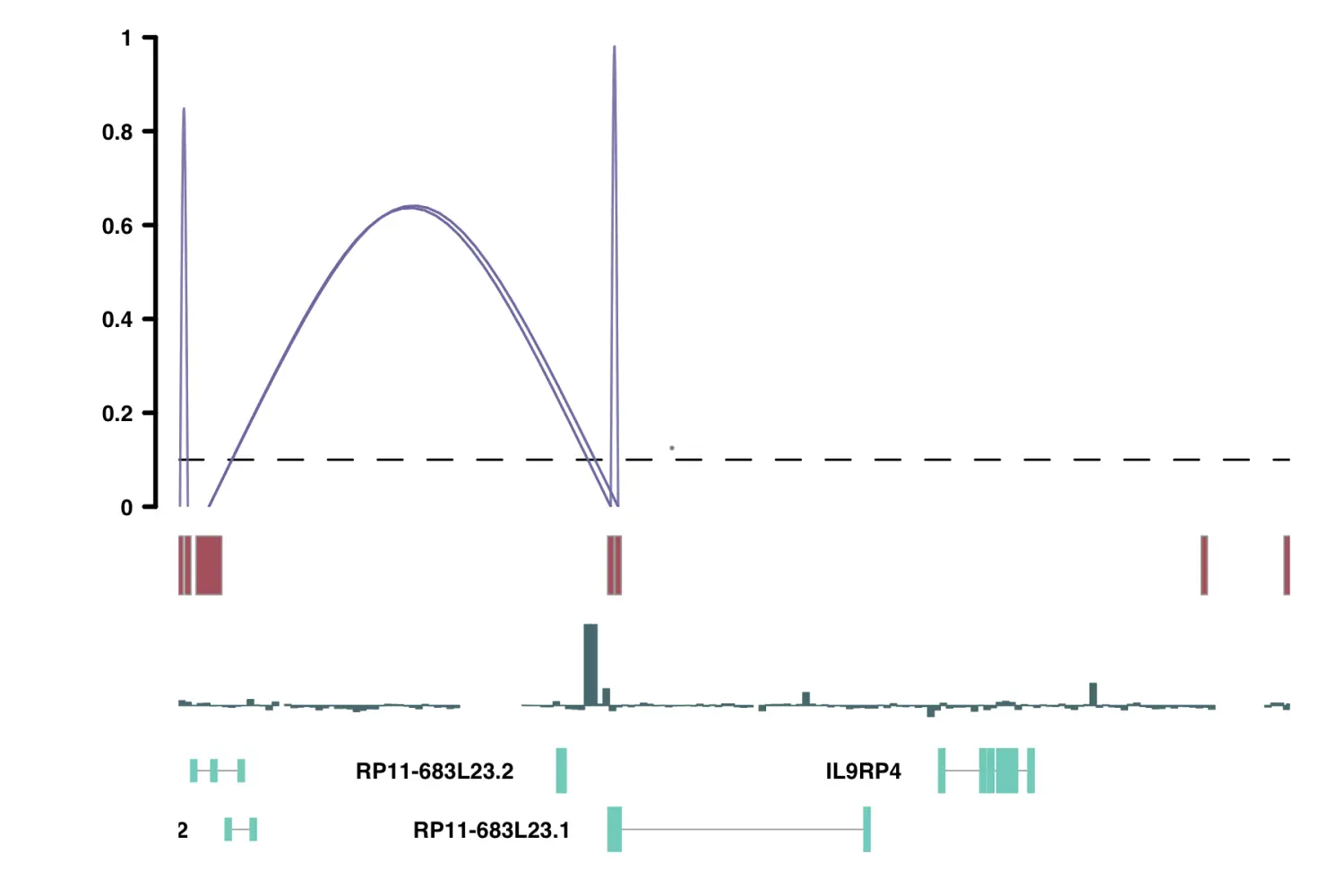
Important Considerations for Non-Human Data
Cicero的默认参数是设计用于人类和小鼠的。对于其他不同的模式生物,我们需要更改一些相应参数,以下是对每个参数的简短说明: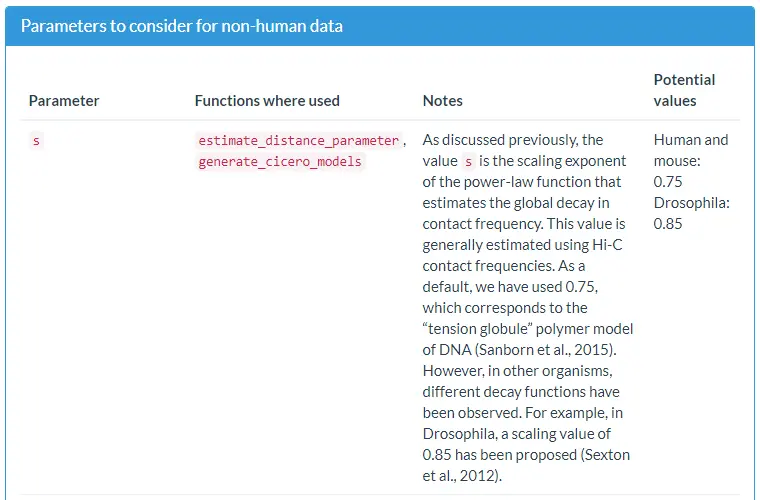

参考来源:https://cole-trapnell-lab.github.io/cicero-release/docs/

若有收获,就点个赞吧
0 人点赞
