(sc)DNA甲基化分析软件
Methylscaper
Methylscaper: an R/shiny app for joint visualization of DNA methylation and nucleosome occupancy in single-molecule and single-cell data
摘要:差异 DNA 甲基化和染色质可及性与疾病发展有关,尤其是癌症。允许在同一反应和单分子或单细胞水平上分析这些表观遗传机制的方法不断出现。然而,挑战在于联合可视化和分析数据的异构性质并提取监管洞察力。在这里,我们提出了methylscaper,一个同时分析DNA甲基化和染色质可及性景观的可视化框架。Methylscaper 实施加权主成分分析,对 DNA 分子进行排序,每个分子都提供一个外等位基因染色质状态的记录,并揭示核小体定位、转录因子占据和 DNA 甲基化的模式。我们展示了methylscaper’ s 用于单个模板 (MAPit-BGS) 数据集和单细胞核小体、甲基化和转录测序 (scNMT-seq) 数据集的长读长单分子甲基转移酶可访问性协议。与其他程序相比,methylscaper 能够轻松识别与转录状态生物学相关的染色质特征,同时扩展到更大的数据集。
可用性和实现: Methylscaper,在 R(版本 > 4.1)中实现,可在
Bioconductor:https://bioconductor.org/packages/methylscaper/;
GitHub:https://github.com/rhondabacher/methylscaper/ ;
Web :https://methylscaper.com;
注:需要DNA甲基化和染色体可及性两种组学的数据
scAI
scAI: an unsupervised approach for the integrative analysis of parallel single-cell transcriptomic and epigenomic profiles
中文标题:一种用于并行单细胞转录组和表观基因组图谱综合分析的无监督方法
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-1932-8
发表时间:2020.02
摘要:同时测量同一单个细胞中的转录组和表观基因组谱,为了解细胞命运提供了前所未有的机会。然而,缺乏对此类数据进行综合分析的有效方法。在这里,我们提出了一种单细胞聚集和整合 (scAI) 方法,从平行转录组和表观基因组谱中解卷积细胞异质性。通过迭代学习,scAI 在以无监督方式学习的相似细胞中聚合稀疏的表观基因组信号,从而实现与转录组测量的相干融合。模拟研究和对三个真实数据集的应用证明了它能够剖析转录组和表观基因组层内的细胞异质性并理解转录调控机制。
methylpy
GitHub:https://github.com/yupenghe/methylpy(流程化工具),和bismark有得一比
Welcome to the home page of methylpy, a pyhton-based analysis pipeline for
- (single-cell) (whole-genome) bisulfite sequencing data
- (single-cell) NOMe-seq data
- differential methylation analysis
methylpy 能做什么分析?
Processing bisulfite sequencing data and NOMe-seq data
快速灵活的单端和双端数据管道
从原始读数 (fastq) 到甲基化状态和/或开放染色质读数
还支持从对齐中获取读数(BAM 文件)
包括读取修剪、质量过滤器和 PCR 重复删除的选项
接受压缩输入并生成压缩输出
支持后亚硫酸氢盐适配器标记 (PBAT) 数据
Calling differentially methylated regions (DMRs)
单胞嘧啶水平的 DMR 调用
支持跨 2 个或多个样本/组的比较
保守而准确
通过组合相邻胞嘧啶的数据来处理低覆盖率数据的有用功能
注:这个软件是人家为了分析发表在Nature的文章顺手开发的一个软件,没想到使用的人还挺多的,真的佩服这些大佬!
Msuite
https://www.szbl.ac.cn/infomation/research/825.html
https://github.com/hellosunking/Msuite
深圳湾实验室孙坤教授课题组:https://icr.szbl.ac.cn/AListOfTeams/info.aspx?itemid=28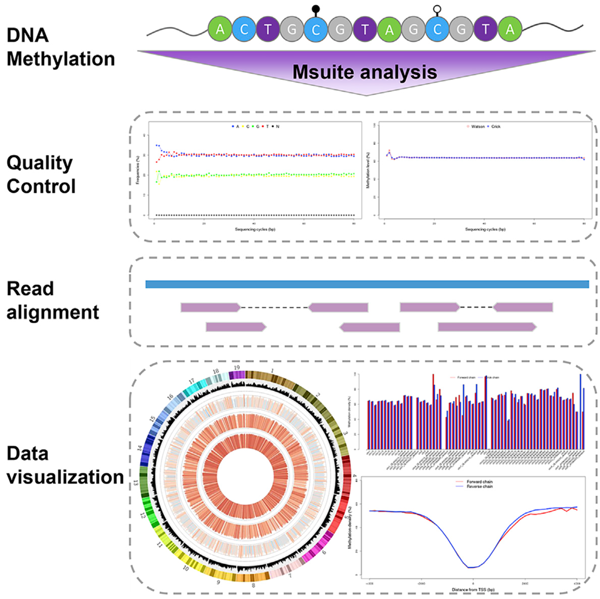
图1. Msuite的工作流程和数据可视化
methySig
GitHub: https://github.com/sartorlab/methylSig
主要功能
- Read data
- Optionally filter data by coverage and/or location
- Optionally aggregate data into regions
- Optionally filter data by coverage in a minimum number of samples per group
- Test for differential methylation
dnameth
Pipelines for Whole Genome and Reduced Representation Bisulfite-seq. Language: Python
Github:https://github.com/databio/dnameth_pipelines
开发软件的这个实验室/研究所好强,记录一下地址:https://databio.org/software/
HISAT-3N
获取地址:
文章标题:Rapid and accurate alignment of nucleotide conversion sequencing reads with HISAT-3N
文章地址:https://genome.cshlp.org/content/early/2021/06/08/gr.275193.120.abstract
比对原理:HISAT-3N 的设计基于HISAT2测序软件,加入了三核酸比对算法(three-nucleotide alignment algorithm)。HISAT-3N首先把reads和reference中所有的C都转化成T,然后用只有三种碱基的reads和只有三种碱基的reference做比对,最后根据原始序列过滤比对结果。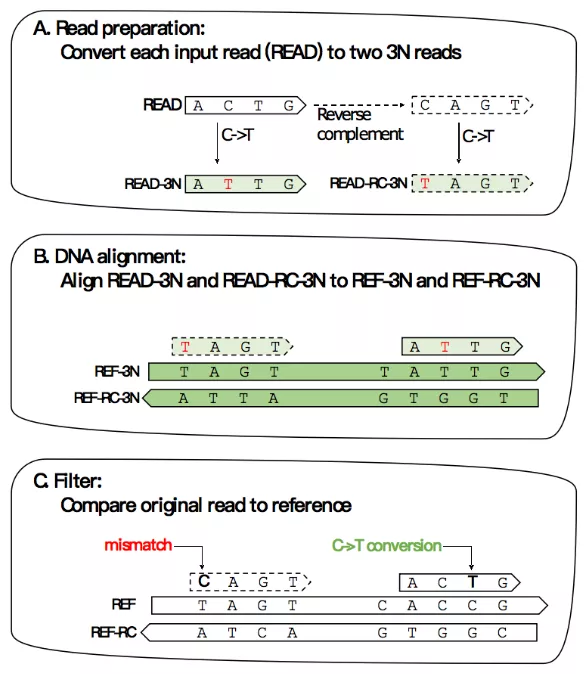
Supplementary Figure 1. Analysis workflow in HISAT-3N for SLAM-seq reads.(A) HISAT-3N converts each input read (READ) to two 3N reads: READ-3N and READ-RC-3N. READ-3N is READ with all
thymine replaced by cytosine. READ-RC-3N is the reverse complement of READ, plus the replacement of cytosine with thymine. (B) HISAT-3N maps the two 3N reads to both REF-3N and REF-RC-3N references using prebuilt indexes. (C) After the three-nucleotide alignment, HISAT-3N compares the original read sequence (READ) to the original fournucleotide references (REF and REF-RC) to identify unmethylated cytosine positions and re-calculate an alignment score accordingly.
methylCtools
这个比对工具来自于2021.03发表在NBT的XRBS技术
文章地址:https://www.nature.com/articles/s41587-021-00910-x#Sec9
MethylStar
GitHub:https://github.com/jlab-code/MethylStar
A fast and robust pre-processing pipeline for bulk or single-cell whole-genome bisulfite sequencing (WGBS) data
文章:https://link.springer.com/article/10.1186/s12864-020-06886-3#Sec13
BMC Genomics,3.59分
简要说明:
To process a large number of WGBS samples in a consistent, documented and reproducible manner it is advisable to use a pipeline system. MethylStar is a fast, stable and flexible pre-processing pipeline for bulk or single-cell (de-multiplexed) WGBS data.
Key features
(1) MethylStar provides a user-friendly interface for experts/non-experts that runs on a Unix based environment.
(2) Offers efficient memory usage and multithreading/multi-core processing support during all pipeline steps.
(3) Better flexibility to the user to adjust parameters, execute and re-execute individual steps.
(4) Generates standard outputs for downstream analysis (formats compatible with DMR-callers such as Methylkit, DMRcaller) and visualisation on genome browsers (bedGraph/BigWig).
Pipeline Steps:质控,比对,评估比对质量,methylation calling。
In its current implementation, MethylStar comprises of the following core NGS components:
(A) Trimmomatic: A flexible read trimming tool (Java based) for processing of raw fastq reads for both single- and paired-end data.
(B) Bismark: Alignment, removal of PCR duplicates and cytosine context extraction steps are performed with the Bismark software suite. Alignments can be performed for both WGBS and Post Bisulfite Adapter tagging (PBAT) approaches for single-cell libraries. Bisulfite treated reads are mapped using the short read aligner Bowtie 2, and therefore it is a requirement that Bowtie 1 or Bowtie 2 are also installed on your machine. Bismark also requires SAMtools to be pre-installed on the computer.
(C) FastQC and bedtools: Tools for assessing data quality.
(D) METHimpute: Cytosine-level methylation calls can be obtained with METHimpute, a Bioconductor package for inferring the methylation status/level of individual cytosines, even in the presence of low sequencing depth and/or missing data.
Note: For information on specific software versions, please refer to Installation and Configuration section.
MethGET
GitHub:https://github.com/Jason-Teng/MethGET
一个用于分析全基因组DNA甲基化和基因表达之间的相关性的网页版工具:https://paoyang.ipmb.sinica.edu.tw/methget。
文章发表在:https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-6722-x
文章中的图即为该软件所能绘制的图形,建议先看下这篇文章再决定要不要考虑这款软件。
MethGET 的输入文件:
CGmap.gz file (need gzip compressed format) is the output of BS-Seeker2.
chr1 G 13538 CG CG 0.6 6 10chr1 G 13539 CHG CC 0.0 0 9chr1 G 13541 CHH CA 0.0 0 9chr1 G 13545 CHH CA 0.0 0 8
其它比对软件获得的methylation calling可以使用软件自带的methcalls2CGmap.py这个python脚本进行文件格式转换为CGmap.gz。
包含以下几个功能模块:
MIRA
依赖语言:R
MIRA:基于 DNA 甲基化推断调节活性的 R 包
摘要: DNA甲基化包含有关细胞调节状态的信息。MIRA 将基因组规模的 DNA 甲基化数据聚合到具有共享生物学注释的给定区域集的 DNA 甲基化谱中。使用此配置文件,MIRA 推断和评分区域集的集体监管活动。MIRA 有助于在经典监管分析难以进行的情况下进行监管分析,并允许利用区域集的公共资源来对 DNA 甲基化数据集的监管状态进行新的洞察。可用性和实施: http://bioconductor.org/packages/MIRA。
scDeconv
scDeconv:一个 R 包,用于对 bulk DNA 甲基化数据与 scRNA-seq 数据和配对的 bulk RNA-DNA 甲基化数据进行反卷积
scDeconv: an R package to deconvolve bulk DNA methylation data with scRNA-seq data and paired bulk RNA–DNA methylation data
Liu, Yu. “scDeconv: an R package to deconvolve bulk DNA methylation data with scRNA-seq data and paired bulk RNA-DNA methylation data.” Briefings in bioinformatics, bbac150. 23 Apr. 2022, doi:10.1093/bib/bbac150
EnsDeconv
Robust and accurate estimation of cellular fraction from tissue omics data via ensemble deconvolution
通过整体反卷积从组织组学数据中稳健而准确地估计细胞分数
Cai, Manqi et al. “Robust and accurate estimation of cellular fraction from tissue omics data via ensemble deconvolution.” Bioinformatics (Oxford, England), btac279. 19 Apr. 2022, doi:10.1093/bioinformatics/btac279
基于测序的甲基化组进行细胞类型预测
EpiDISH
A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies
GitHub:https://github.com/sjczheng/EpiDISH
文章地址:https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-017-1511-5
注:目前软件只适用于甲基化芯片数据,但作者在GitHub 的一个issues说了可以通过修改一下WGBS或RRBS的位点甲基化水平矩阵,也可以作为EpiDISH 的输入。
软件使用示例:https://www.cnblogs.com/chenwenyan/p/14589958.html
使用了该软件进行分析的文章:Meta-analysis of genome-wide DNA methylation identifies shared associations across neurodegenerative disorders
更多参考资料:
- 反卷积pipeline评估群体甲基化测序中的细胞类型
英文标题:Systematic evaluation of cell-type deconvolution pipelines for sequencing-based bulk DNA methylomes
中文翻译:基于测序的群体DNA甲基组的细胞类型反卷积管道的系统评价
2021.12刚发在预印本的论文
与其他组学联合分析
MethylationToActivity(M2A)
原文链接:MethylationToActivity: a deep-learning framework that reveals promoter activity landscapes from DNA methylomes in individual tumors
GitHub:https://github.com/chenlab-sj/M2A
简单介绍:
MethylationToActivity (M2A)是一个机器学习框架,使用卷积神经网络(CNN)从全基因组亚硫酸氢盐测序(WGBS)中推断组蛋白修饰(histone modification)富集情况。到目前为止,从采用 M-values(M-value=log2(Beta/1-Beta))的制表符分隔文本文件格式,WGBS的 H3K27ac 和 H3K4me3 富集预测都得到了支持。
该研究通过卷积神经网络(convolutional neural network)来提取启动子及其相邻区域的DNA甲基化高维特征(涵盖启动子及其相邻区域的DNA甲基化特征),最后通过这些特征变量来建立一个模型,用于推断全基因组启动子(H3K4me3和H3K27ac)以及基因转录(FPKM from RNA-seq)的活性。
M2A 由以下五个步骤组成,包括迁移学习(including transfer-learning):
| Process | Description |
|---|---|
| 1_ResponseVariable | Generate histone enrichment for each unique promote region (transfer-learning only) |
| 2_MethylationFeatures | Process WGBS features for model input |
| 3_CombineInput | Scale and recombine features, and for transfer learning, calculated HM values |
| 4_TransferLearning | Train fully-connected layers of a particular model for increased performance in your domain of interest (Optional) |
| 5_RunModel | Using pre-generated input, get HM predictions for each unique promoter region |
输入要求:
M2A requires five inputs, defined in a YAML file as CWL inputs. E.g., inputs.yml:
chipBigwig:class: Filepath: sample.bwinputBigwig:class: Filepath: input.bwcurated:class: Filepath: sites.txtpromoterDefinitions:class: Filepath: promoters.txtmodel:class: Filepath: model.h5
每种输入的描述:
| Name | Description |
|---|---|
| Sample HM bigwig file (only if using M2A with Transfer) | HM ChIP-seq experiment bigwig track. |
| Sample HM control (Input) bigwig (only if using M2A with Transfer) | ChIP-seq Experiment control (Input) bigwig track. |
| WGBS data file | M-values by chromosome and position (non-standard format, see below). |
| Promoter region definition file (provided, or user defined) | File describing promoter regions to be predicted. (non-standard format, see below) |
| Model weights (provided, or user defined from transfer) | hdf5 model weights for either H3K27ac prediction OR H3K4me3 prediction |
具体每种输入的文件格式见GitHub的说明文档。
iNMF框架整合分析单细胞多组学数据

Iterative single-cell multi-omic integration using online learning
https://www.nature.com/articles/s41587-021-00867-x
摘要:整合大型单细胞基因表达、染色质可及性和 DNA 甲基化数据集需要通用且可扩展的计算方法。在这里,我们描述了在线综合非负矩阵分解 (iNMF),这是一种用于集成大型、多样化和不断到达的单细胞数据集的算法。我们的方法可以扩展到使用固定内存的任意大量单元格,在生成新数据集时迭代地合并它们,并允许许多用户通过互联网流式传输大数据集的单个副本来同时分析它。迭代数据添加也可用于将新数据映射到参考数据集。与先前方法的比较表明,效率的提高不会牺牲数据集对齐和集群保存性能。
交互式/网页版
SMART App
The SMART App: an interactive web application for comprehensive DNA methylation analysis and visualization
SMART 应用程序可从 http://www.bioinfo-zs.com/smartapp 获得。
背景: 基于癌症基因组图谱 (TCGA) 的数据挖掘极大地促进了癌症基因组研究,并为癌症研究人员提供了前所未有的机会。然而,现有的用于 DNA 甲基化分析的网络应用程序并不能充分满足实验生物学家的需求,而且往往需要许多附加功能。
结果: 为了便于 DNA 甲基化分析,我们推出了 SMART(Shiny Methylation Analysis Resource Tool)应用程序,用于全面分析 TCGA 项目的 DNA 甲基化数据。SMART App将多组学和临床数据与DNA甲基化相结合,提供CpG可视化、泛癌甲基化谱、差异甲基化分析、相关性分析和生存分析等关键交互和定制功能,供用户分析不同癌症类型的DNA甲基化以多维方式。
结论: SMART App 为用户,特别是没有编程背景的湿实验科学家提供了一种新的方法来分析科学大数据并促进数据挖掘。
软件的介绍:SMART App:DNA甲基化综合分析网页工具介绍
我的想法:因为是基于TCGA数据库,其分析功能应该都是基于DNAmeth array开发的,不知道能不能应用于BS-seq等高通量测序数据的分析。
初次使用发现比较卡,使用基因TP53进行测试,图形效果如下
scmeth (R package)
文章标题:A (fire)cloud-based DNA methylation data preprocessing and quality control platform
Documentation for this pipeline and all the workflows can be accessed at http://aryee.mgh.harvard.edu/dna-methylation-tools/.
scmeth is available through the Bioconductor project (https://www.bioconductor.org/packages/release/bioc/html/scmeth.html)..)
文章介绍的是一个DNA甲基化数据预处理和质量控制的云平台,或者说是一组数据预处理的pipeline。这个pipeline包括一个新开发的R包scmeth。它可以用于对大型数据集进行一系列有效的QC分析,例如单细胞分析中常见的数据集。
QC 报告可用于识别低质量批次或样品,并提供指标,包括读取次数、总 CpG 覆盖率、亚硫酸氢盐转化率、甲基化分布、基因组特征覆盖率(例如启动子、增强子)、下采样饱和曲线和甲基化分布。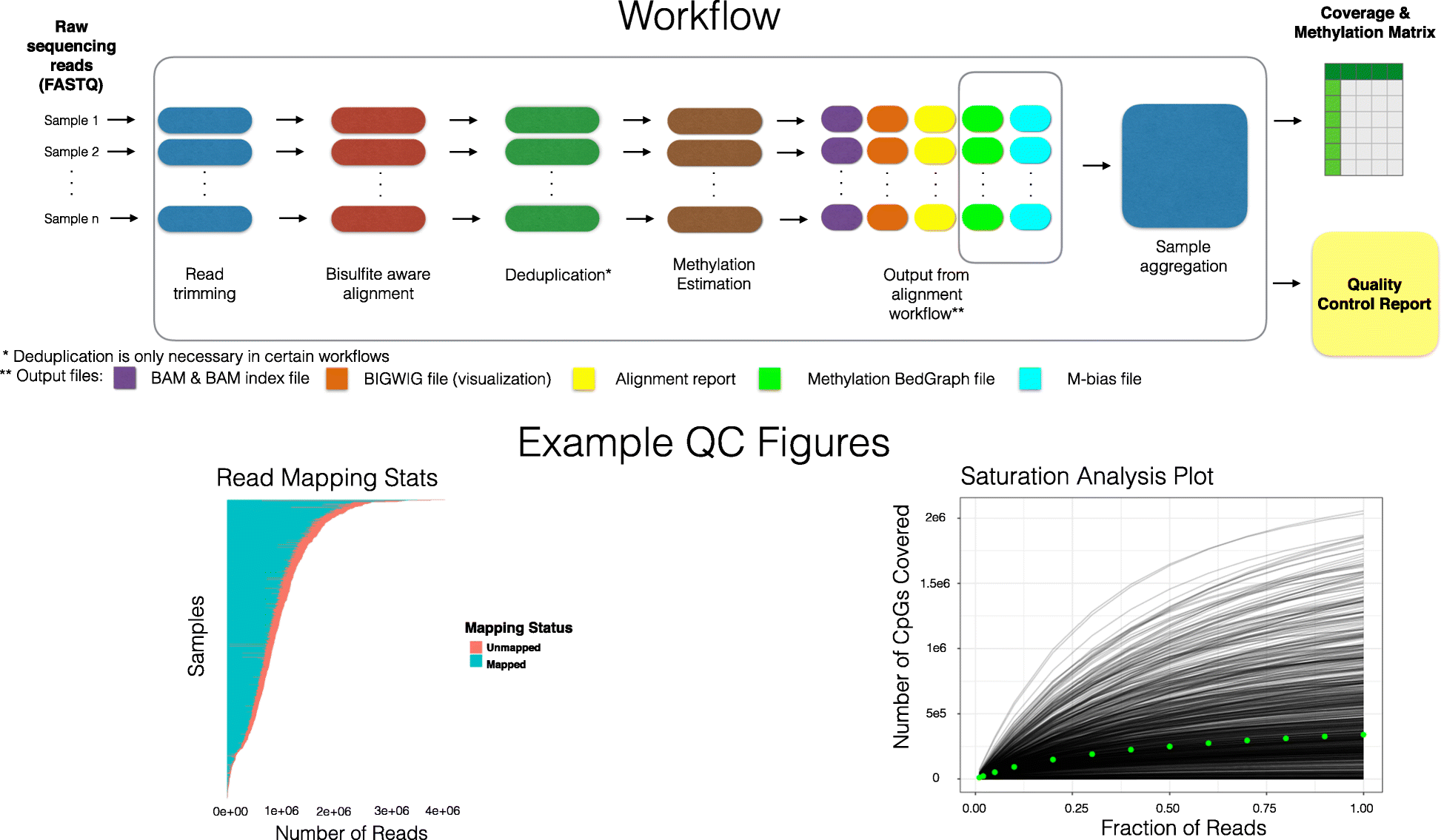
甲基化分析工作流程概述。原始测序序列 (FASTQ) 文件,首先通过每个样品的质控、比对和预处理步骤进行处理,然后是将所有样品的数据组合成矩阵格式并生成 QC 报告的聚合步骤。

若有收获,就点个赞吧
0 人点赞
