背景知识
什么是批次效应?
批次效应是指样品在不同批次中处理和测量产生的与试验期间记录的任何生物变异无关的技术差异。
去除基因表达量批次效应的主要方法有removeBatchEffect(limma包)、ComBat方法(sva包)、替代变量分析法、距离加权判别法和基于比值的方法等,
注意事项:
- 批次效应尽量从实验上避免,不论是单细胞还是群体组学测序数据,在进行批次效应校正时,软件是无法区分批次带来的影响和真实的 biological different!
- 批次效应不能被消除,只有尽可能的降低;
- 批次因素和分组因素可能重叠,所以直接对原数据矫正批次可能会抵消一部分真实生物学因素;
- 使用removeBatchEffect或者ComBat函数后得到的表达数据,仅可用于衔接可视化(如聚类、PCA等),可视化展示;不能使用批次校正后的数据进行差异分析!(待证实)
- 如果想要在鉴定差异基因的过程中降低批次效应,将批次加入到design中。即使用公式:
disign = ~ batch + groups。
去除/减弱批次效应的方法
methylKit包的removeComp
函数:assocComp + removeComp
library(methylKit)data(methylKit)sampleAnnotation=data.frame(batch_id=c("a","a","b","b"),age=c(19,34,23,40))as=assocComp(mBase=methylBase.obj,sampleAnnotation)as# $pcs# PC1 PC2 PC3 PC4# test1 -0.4978699 -0.5220504 0.68923849 -0.06737363# test2 -0.4990924 -0.4805506 -0.71827964 0.06365693# ctrl1 -0.5016543 0.4938800 0.08068700 0.70563101# ctrl2 -0.5013734 0.5026102 -0.05014261 -0.70249091## $vars# [1] 92.271885 4.525328 1.870950 1.331837## $association# PC1 PC2 PC3 PC4# batch_id 0.3333333 0.3333333 1.0000000 1.0000000# age 0.5864358 0.6794346 0.3140251 0.3467957# construct a new object by removing the first pricipal component# from percent methylation value matrixnewObj=removeComp(methylBase.obj,comp=1)mat=percMethylation(methylBase.obj)head(mat)newmat=percMethylation(newObj)head(newmat)
Tips:
- 输入对象为methylKit包对象且不允许存在NA。
输入的methylBase.obj对象是methylKit包的对象,其格式如下图: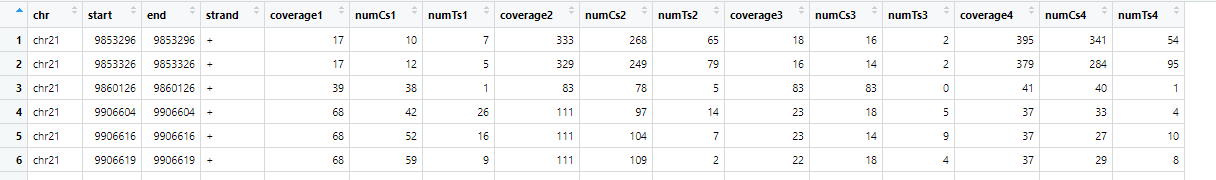
但是有一点需要注意,assocComp 内部是涉及主成分分析的,所以assocComp 函数不允许输入的对象矩阵存在NA,别人也可能遇到同样的描述:Error in svd(x, nu = 0, nv = k) : infinite or missing values in ‘x’
- 该不该移除批次效应?
未移除批次效应与移除部分批次效应的矩阵的前后比较,看起来这几个位点甲基化水平都是降低了,不知道理论上是不是这样子的,所以要不要进行批次效应的去除?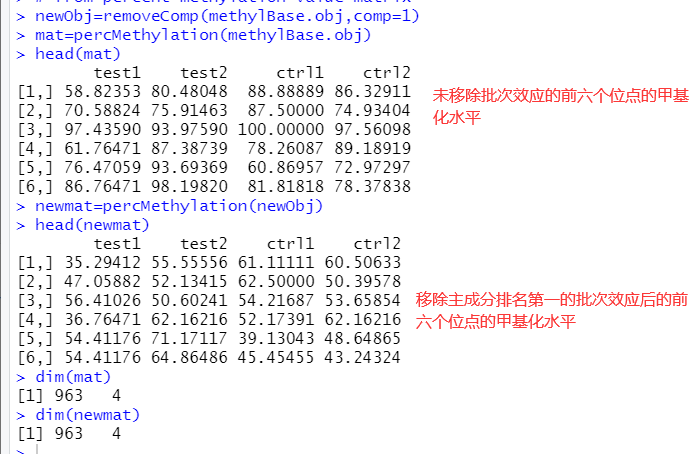
参考:
- https://bioconductor.org/packages/release/bioc/vignettes/methylKit/inst/doc/methylKit.html#34_Batch_effects
- https://www.rdocumentation.org/packages/methylKit/versions/0.99.2/topics/assocComp
sva包的combat
北京大学李程课题组开发的sva包的ComBat函数.
ComBat是基于经典贝叶斯的分析方法,运用已知的批次信息对高通量数据进行批次校正。在sva R package 中提供了ComBat用于处理批次效应。ComBat有两个方法可供选择,一种是基于参数和一种非参数方法,combat函数的par.prior参数可以设置。函数输入数据为经过标准化的数据矩阵,返回结果为经过批次校正后的一个数据矩阵。
示例代码一:基于bladderbatch包的示例数据
library(BiocManager)BiocManager::install("sva")BiocManager::install("bladderbatch")library(sva)library(bladderbatch)data(bladderdata)dat <- bladderEset[1:50,]pheno = pData(dat)edata = exprs(dat)batch = pheno$batchmod = model.matrix(~as.factor(cancer), data=pheno)# > head(pheno)# sample outcome batch cancer# GSM71019.CEL 1 Normal 3 Normal# GSM71020.CEL 2 Normal 2 Normal# GSM71021.CEL 3 Normal 2 Normal# GSM71022.CEL 4 Normal 3 Normalhead(edata)# GSM71019.CEL GSM71020.CEL GSM71021.CEL# 1007_s_at 10.115170 8.628044 8.779235# 1053_at 5.345168 5.063598 5.113116# 117_at 6.348024 6.663625 6.465892# > head(mod)# (Intercept)# GSM71019.CEL 1# GSM71020.CEL 1# GSM71021.CEL 1# GSM71022.CEL 1# parametric adjustmentcombat_edata1 = ComBat(dat=edata, batch=batch, mod=NULL, par.prior=TRUE, prior.plots=FALSE)# non-parametric adjustment, mean-only versioncombat_edata2 = ComBat(dat=edata, batch=batch, mod=NULL, par.prior=FALSE, mean.only=TRUE)# reference-batch version, with covariatescombat_edata3 = ComBat(dat=edata, batch=batch, mod=mod, par.prior=TRUE, ref.batch=3)
另外,ber包同样可以校正批次效应。同样运用上面的bladder cancer数据。在这个R包中提供了六个函数来进行批次校正,ber函数,ber_bg, combat_np, combat_p,mean_centering,standardization函数。对于这六个函数来说,输入数据行对应的是样本,列对应的是变量。
dat = t(edata)class = data.frame(pheno$cancer)batch = as.factor(pheno$batch)ber_edata1 = ber(dat, batch, class)ber_edata2 = ber_bg(dat, batch, class)ber_edata3 = combat_np(dat, batch, class)ber_edata4 = combat_p(dat, batch, class)
示例代码二:
摘抄于:sva ComBat去除GEO数据批次效应
# 加载R包和GEO示例的转录组表达数据BiocManager::install("sva")BiocManager::install("bladderbatch")library(sva)library(bladderbatch)load("GeoData.rda")#原始数据FPKM[1:5,1:5]GSE102746_UC_1 GSE102746_UC_2 GSE102746_UC_3 GSE102746_UC_4 GSE102746_UC_5ENSG00000008300 0.795651 12.74320 12.8748 0.296484 1.70187ENSG00000044574 83.731600 167.95100 29.3973 85.368700 49.98990ENSG00000067082 78.742500 130.74000 64.4235 68.536000 80.07060ENSG00000071575 9.407460 8.30127 30.0466 9.541390 7.68865ENSG00000089159 29.575000 59.32220 17.5021 27.272200 22.99260#批次信息sample_infor[1:5,]sample_name GSE_name Batch_GSE data_type diseaseGSE102746_UC_1 GSE102746_UC_1 GSE102746 1 test_data UCGSE102746_UC_2 GSE102746_UC_2 GSE102746 1 test_data UCGSE102746_UC_3 GSE102746_UC_3 GSE102746 1 test_data UCGSE102746_UC_4 GSE102746_UC_4 GSE102746 1 test_data UCGSE102746_UC_5 GSE102746_UC_5 GSE102746 1 test_data UClibrary("FactoMineR")library("factoextra")pca.plot = function(dat,col){df.pca <- PCA(t(dat), graph = FALSE)fviz_pca_ind(df.pca,geom.ind = "point",col.ind = col ,addEllipses = TRUE,legend.title = "Groups")}pca.plot(FPKM,factor(sample_infor$GSE_name))
校正前的PCA显示,不同数据集区分明显,可能是批次效应影响较大。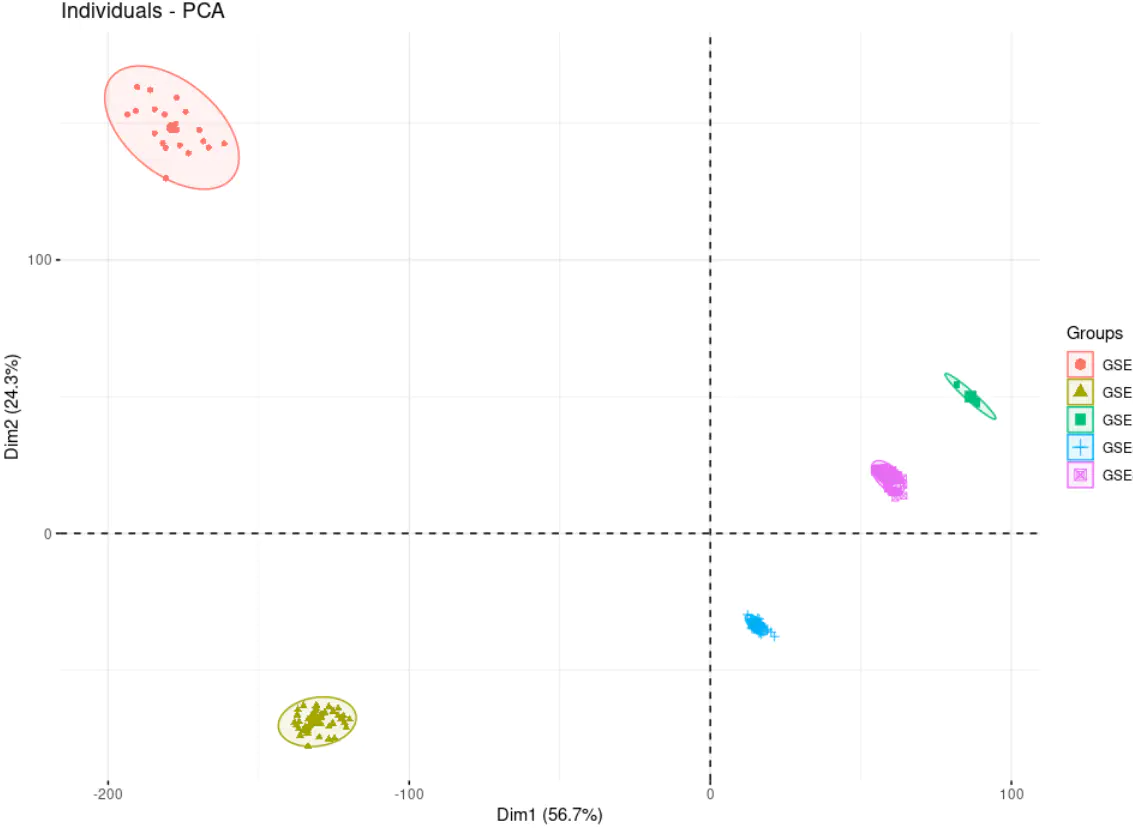
使用combat函数进行批次效应校正:
#设置model(可选)mod = model.matrix(~as.factor(disease), data=sample_infor)#校正其实就一步combat_FPKM <- ComBat(dat = as.matrix(log2(FPKM+1)), batch = sample_infor$Batch_GSE,mod = mod,par.prior = F)pca.plot(combat_FPKM,factor(sample_infor$GSE_name))
校正前后具有明显差异: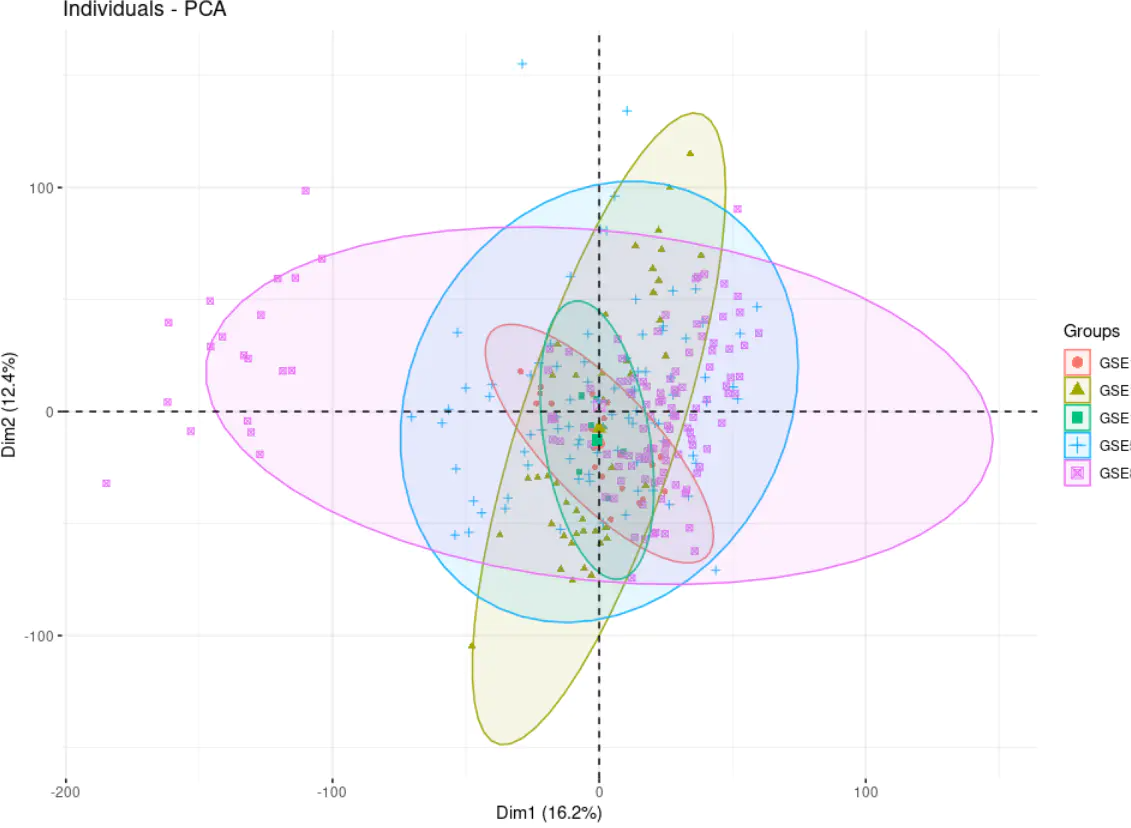
报错记录
- 输入矩阵内存在NA

这种情况是因为矩阵内存在NA值/缺失值,这种情况去除矩阵内所有包含缺失值的行或列即可。
比如使用na.omit函数。
edata <- na.omit(edata)table(is.na(edata))
limma包的removeBatchEffect
摘抄自:生信宝典:高通量数据中批次效应的鉴定和处理 - 系列总结和更新
如果批次信息有多个或者不是分组变量而是类似SVA预测出的数值混杂因素,则需使用limma的removeBatchEffect(这里使用的是SVA预测出的全部3个混杂因素进行的校正。)。
样品在PC1和PC2组成的空间的分布与ComBat结果类似,只是PC1能解释的差异略小一些。
基本使用方法:
SV = metadata[,c("SV1","SV2","SV3")]expr_mat_batch_correct_limma1 <- removeBatchEffect(expr_mat, covariates = SV, design=modcombat)sp_pca(expr_mat_batch_correct_limma1[1:5000,], metadata,color_variable="conditions", shape_variable = "individual") +aes(size=1) + guides(size = F)
参考资料
若有侵权请留言或联系本人。

若有收获,就点个赞吧
0 人点赞
