每周从“文献鸟”获取与DNA甲基化或单细胞相关的文献,其中挑选相关性较高的几篇进行导读。
第一周
EpiScanpy: integrated single-cell epigenomic analysis
中文标题:EpiScanpy:整合单细胞表观基因组分析
发表时间:2021.09.01;Nature Communications
Github:https://github.com/colomemaria/epiScanpy,基于python3开发,是热门的单细胞转录组数据分析工具scanpy在表观领域的扩展;
Document: https://colomemaria.github.io/episcanpy_doc/
摘要:EpiScanpy 是一个用于分析单细胞表观基因组数据的工具包,即单细胞 DNA 甲基化和单细胞 ATAC-seq 数据。 为了解决表观基因组学数据中的模态特定挑战,epiScanpy 使用多个特征空间结构量化表观基因组,并使用细胞之间的表观基因组距离构建最近邻图。 EpiScanpy 使许多现有的 scRNA-seq 工作流程可用,从 scanpy 到来自其他组学模式的大规模单细胞数据,包括用于常见聚类、降维、细胞类型识别和轨迹学习技术的方法,以及图谱集成工具 用于 scATAC-seq 数据集。 该工具包还具有许多有用的下游功能,例如差异甲基化和差异开放性调用、将感兴趣的表观基因组特征映射到其最近的基因,或使用染色质开放性构建基因活性矩阵。 我们成功地对 EpiScanpy 与其他 scATAC-seq 分析工具进行了基准测试,并展示了其在区分细胞类型方面的出色表现。
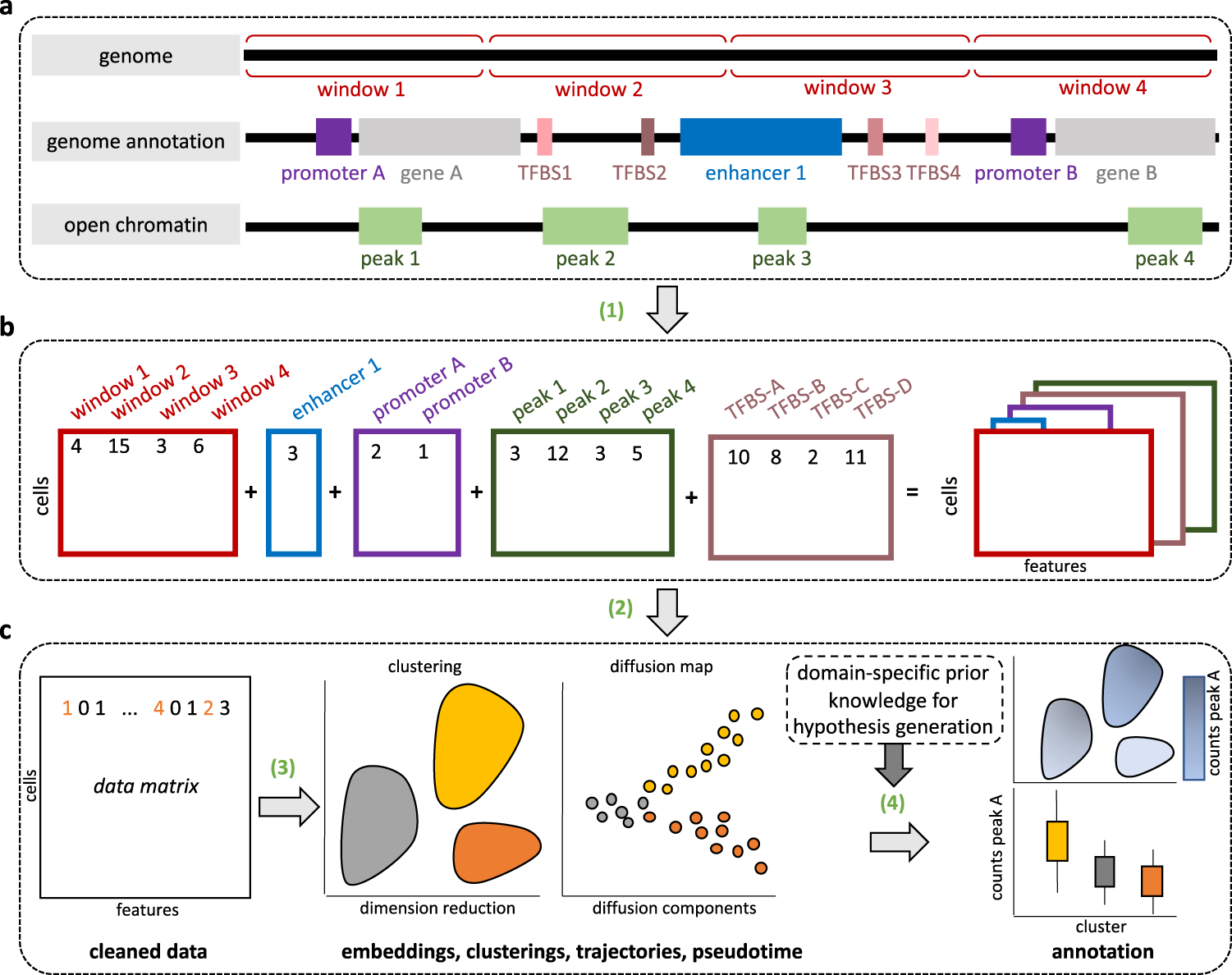
图1. EpiScanpy 工作流程图
a. EpiScanpy 量化不同基因组区域组的染色质开放度和 DNA 甲基化,(箭头,步骤1) ;
b. 构建具有reads counts(scATAC-seq)或 DNA 甲基化水平(单细胞 DNA 甲基化)的计数矩阵 ;
c. 在数据预处理 (步骤2) 之后,应用无监督学习算法 (集群、轨迹、谱系树) (步骤3)。 差异开放性和甲基化调用允许识别标记基因座,可用于细胞类型和谱系树识别 (步骤4)。
引用文献的12, 13也是用于分析单细胞甲基化组的软件
示例教程
Episcanpy Tutorials:https://colomemaria.github.io/episcanpy_doc/examples.html
在这里我们只对 Single cell DNA methylation 的分析教程进行复现,Single cell ATAC-seq的教程暂时不复现了。
构建计数矩阵
一、输入文件
- methylation calling
第一个教程展示了如何为不同胞嘧啶上下文中的不同特征空间(窗口、启动子)构建计数矩阵。
假如比对软件使用的是bismark,methylation calling使用bismark_methylation_extractor命令,那么我们一般能拿到以下三种格式的methylation calling 文件。
Generating additional output in bedGraph and coverage formatformat1. bedGraph format: <Chromosome> <Start Position> <End Position> <Methylation Percentage>1 191491 191491 1001 191507 191507 1001 631788 631788 0format2. coverage format: <Chromosome> <Start Position> <End Position> <Methylation Percentage> <count methylated> <count non-methylated>1 191491 191491 100 3 01 191507 191507 100 3 01 631788 631788 0 0 1format3. comprehensive genome-wide cytosine report(output format: <Chromosome> <Position> <Strand> <count methylated> <count non-methylated> <C-context> <trinucleotide context> )1 10469 + 0 0 CG CGC1 10470 - 0 0 CG CGA1 10471 + 0 0 CG CGG
从教程给出的示例数据下载地址:https://drive.google.com/drive/folders/1TnaVihYDPQ5IUZP3ZQlv7hQIwJVsWnUn,下载了”cell1.tsv”发现其格式如下
chr pos strand mc_class mc_count total methylated1 3003252 + CCT 0 1 01 3003253 + CTG 0 2 01 3003266 + CTA 0 5 0
- 样本配置文件
样本信息表的内容(mouse_annot_5cells_Luo17.csv)
| Sample | Animal age | FACS date | Brain area | Laminar layer | Labeling | FACS channel | FACS count | Bisulfite conversion method | Library type | Library pool | Index i5 | Index i7 | i5 sequence | i7 sequence | random primer index | random primer index sequence | Sequencing run mode | Total reads | Mapped reads | % Mapped reads | Filtered reads | % Filtered reads | mCCC/CCC | mCG/CG | mCH/CH | Estimated mCG/CG | Estimated mCH/CH | Coverage (%) | Neuron type | tSNE x coordinate | tSNE y coordinate |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pool_161_AD002_indexed | 8wk | 22.06.16 | Frontal Cortex | Superficial | Anti-NeuN | 488+ | 1 | Zymo EZ-96 direct | snmC-seq | B1B2 | D501 | D709 | TATAGCCT | CGGCTATG | AD002 | CGATGT | PE150 | 7131590 | 2457414 | 34,50% | 1746951 | 71,10% | 0,00703 | 0,73239 | 0,02246 | 0,7305 | 0,01554 | 4,32 | mL4 | -632,949 | -572,515 |
| Pool_161_AD006_indexed | 8wk | 22.06.16 | Frontal Cortex | Superficial | Anti-NeuN | 488+ | 1 | Zymo EZ-96 direct | snmC-seq | B1B2 | D501 | D709 | TATAGCCT | CGGCTATG | AD006 | GCCAAT | PE150 | 7022858 | 3552348 | 50,60% | 2477680 | 69,70% | 0,00522 | 0,72835 | 0,01918 | 0,72692 | 0,01403 | 6,02 | mL2/3 | -135,826 | -0,0183373 |
| Pool_162_AD002_indexed | 8wk | 22.06.16 | Frontal Cortex | Superficial | Anti-NeuN | 488+ | 1 | Zymo EZ-96 direct | snmC-seq | B1B2 | D502 | D709 | ATAGAGGC | CGGCTATG | AD002 | CGATGT | PE150 | 2958836 | 1413795 | 47,80% | 1109054 | 78,40% | 0,00602 | 0,77029 | 0,02289 | 0,7689 | 0,01697 | 2,88 | mVip | -0,0493594 | 126,162 |
| Pool_162_AD006_indexed | 8wk | 22.06.16 | Frontal Cortex | Superficial | Anti-NeuN | 488+ | 1 | Zymo EZ-96 direct | snmC-seq | B1B2 | D502 | D709 | ATAGAGGC | CGGCTATG | AD006 | GCCAAT | PE150 | 4370610 | 2162353 | 49,50% | 1677114 | 77,60% | 0,00592 | 0,74233 | 0,02241 | 0,7408 | 0,01659 | 4,33 | mNdnf-1 | -331,483 | 115,906 |
| Pool_163_AD006_indexed | 8wk | 22.06.16 | Frontal Cortex | Superficial | Anti-NeuN | 488+ | 1 | Zymo EZ-96 direct | snmC-seq | B1B2 | D503 | D709 | CCTATCCT | CGGCTATG | AD006 | GCCAAT | PE150 | 5319826 | 2771225 | 52,10% | 2048069 | 73,90% | 0,00558 | 0,73922 | 0,02064 | 0,73776 | 0,01514 | 5,21 | mL2/3 | -144,477 | -480,087 |
如果需要可视化好看的,能根据标签进行细胞亚群区分,就必须构建这么一个类似的样本信息表,内容不一定完全相同。
另外的一些文献
1. 脑膜瘤临床适用的综合分子分类
A clinically applicable integrative molecular classification of meningiomas
Nature
2021.08.25
获取地址:https://www.nature.com/articles/s41586-021-03850-3
脑膜瘤是成人中最常见的原发性颅内肿瘤。由于没有有效的药物疗法,出现症状的患者通常接受手术治疗。世界卫生组织对肿瘤的组织病理学分级和手术切除范围(辛普森分级)与疾病的复发有关;然而,它们并不能准确反映所有脑膜瘤的临床表现. 需要可靠地反映肿瘤行为并为治疗提供信息的脑膜瘤的分子分类。在这里,我们通过在统一分析中结合 DNA 体细胞拷贝数畸变、DNA 体细胞点突变、DNA 甲基化和信使 RNA 丰度,介绍了脑膜瘤的四个共有分子组。与现有的分类方案相比,这些分子组更准确地预测了临床结果。每个分子群都表现出独特的原型生物学(免疫原性、良性 NF2野生型、代谢亢进和增殖),为治疗选择提供信息。蛋白质基因组学表征增强了新定义的分子群的稳健性,并发现了我们使用免疫组织化学验证的高度丰富和群特异性的蛋白质靶标。单细胞 RNA 测序揭示了脑膜瘤的个体间差异以及肿瘤细胞内在表达程序的差异,这些差异反映了所识别分子群的生物学特性。
121 名患者的脑膜瘤样本
独立队列的 80 份样本
我们对种系多态性、体细胞点突变和体细胞拷贝数改变进行了全外显子组测序;用于 DNA 甲基化分析的 EPIC芯片分析和 mRNA 测序,用于对发现队列中的所有 121 个肿瘤进行转录组分析。对这些肿瘤中的 96 个进行了全细胞蛋白质组学(图1a)。还对五个健康脑膜样本进行了 DNA 甲基化,以进行甲基化组比较。通过单核 RNA 测序对八个肿瘤和两个健康脑膜样本进行分析,以检查肿瘤内的异质性。
补充图8. 健康脑膜和脑膜瘤之间全基因组 DNA 甲基化模式的差异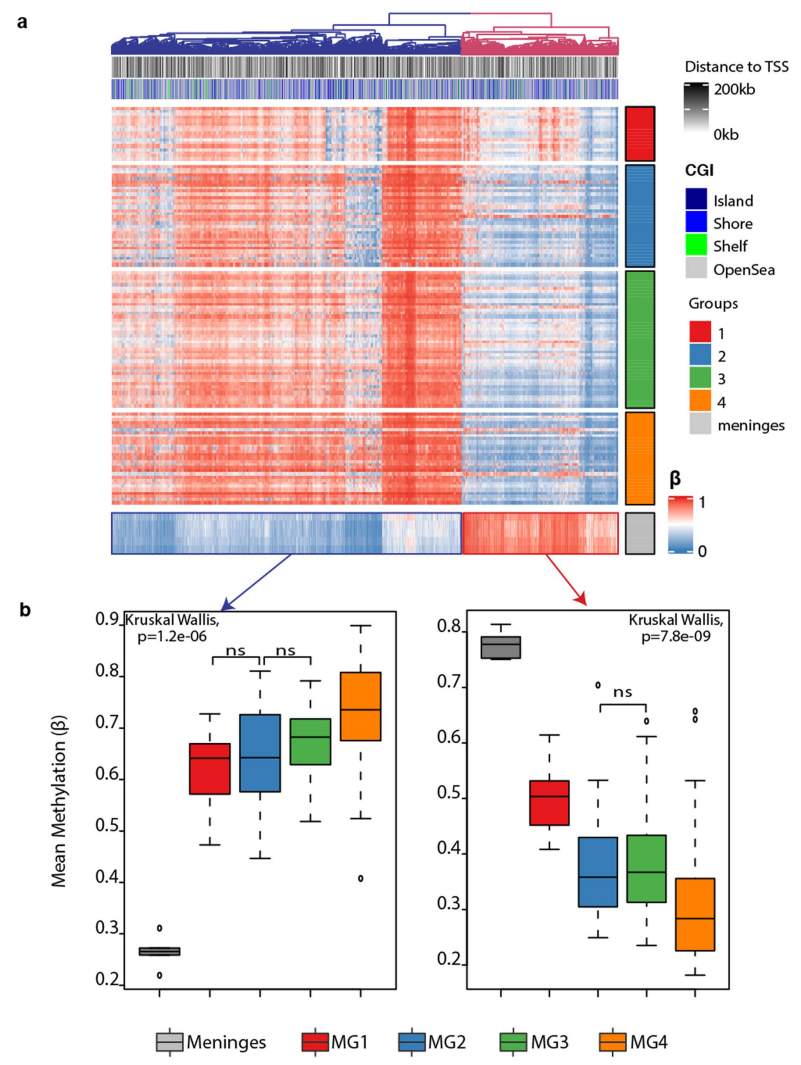
我们鉴定了两组将健康脑膜与整个脑膜瘤区分开来的探针(补充图8a)。在一组中,探针在健康脑膜中完全低甲基化,并逐渐在分子组中获得甲基化,而在另一组中,探针在健康脑膜中完全高甲基化,并在分子组中逐渐失去甲基化。(补充图8b)。
结论:为脑膜瘤社区提供了一个资源,其中包含匹配的多维批量和单细胞分子以及高质量的临床数据。通过在统一分析中整合多种数据类型,我们定义了脑膜瘤的分子分类法,它可以取代现有的分子和临床使用的分类,并有可能为公认的分级方案。
2.隐精子症全基因组甲基化
从隐性男性的睾丸生殖细胞的全基因组甲基化分析指出,反复和功能相关的DNA甲基化变化|
Whole-genome methylation analysis of testicular germ cells from cryptozoospermic men points to recurrent and functionally relevant DNA methylation changes
2021.08.21
Clin Epigenetics (impact factor: 6.6) 2 区
为了找出这些患者的睾丸生殖细胞 (TGC) 是否可能携带异常的 DNA 甲基化,我们比较了四名隐精子症 (CZ) 男性和四名具有正常精子发生并作为对照 (CTR) 的阻塞性无精子症男性的 TGC 甲基化组.
结果
在全基因组水平或 CZ 和 CTR 样本之间的印记区域的DNA 甲基化没有差异。然而,使用严格的过滤器来识别特定组的甲基化差异,我们检测到 271 个差异甲基化区域 (DMR),其中 238 个在 CZ 中被高甲基化(二项式检验,p < 2.2 × 10 –16)。DMRs 富含远端调节元件并与 132 个基因相关,其中 61 个基因在精子发生的不同阶段差异表达。与 61 个基因相关的 67 个 DMR 几乎全部 (94%) 在 CZ 中都被高甲基化。
根据单细胞 RNA 测序判断,主要在减数分裂和精子发生过程中表达的 13 个 DMR 相关基因在 CZ 患者中显示出显着不同的表达模式。在其中四个基因中,CZ 男性的启动子高度甲基化,这与这些患者中的表达水平较低有关。在其他九个基因中,其中八个在 CZ 中下调,生殖细胞特异性增强子可能受到影响。
CTR-CZ DMR 与 132 个基因相关。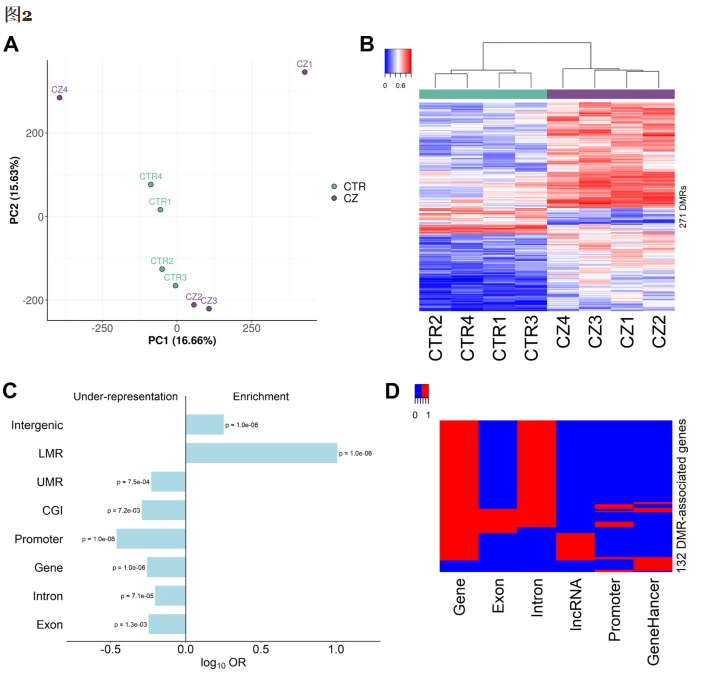
a. 来自对照 (CTR) 和隐精子 (CZ) 睾丸生殖细胞的甲基化组的PCA分群图。2000 万个 CpG 基因座,其中所有样品都显示甲基化值。仅考虑所有样本中最小覆盖率为 5 且最小映射质量为 10 的位点。CTR 睾丸生殖细胞样本为青色,CZ 样本为紫色。
b. 8 个 TGC 样品中 271 个 CTR-CZ DMR 的甲基化值的聚类分析。
c. 功能基因组区域 DMR 的富集/降低。LMR,低甲基化区域;UMR,未甲基化区域;CGI, CpG 岛。
d. DMR 通过重叠基因、启动子和/或“双精英”增强子 (GeneHancer) 与 132 个基因相关联
结论
来自精子发生受损男性的 TGC 在特定基因组区域的 DNA 甲基化水平上与对照 TGC 不同,其中许多似乎是基因调控元件。我们不知道 CZ 男性异常甲基化模式的原因,但一种可能性是观察到的甲基化变化介导或反映了涉及细胞水平基因调控回路的基因表达变化。大多数 DMR 在 CZ 中高度甲基化的事实表明在精子发生过程中上调重要基因的失败。我们假设所描述的 DNA 甲基化变化可能反映或导致精子发生的过早流产,因此不会出现在成熟的活动精子中。
第二周
- DNA甲基化是维持DNA复制时间精度和3D基因组组织完整性所必需的
标题:DNA methylation is required to maintain both DNA replication timing precision and 3D genome organization integrity
Du Q, Smith GC, Luu PL, et al. DNA methylation is required to maintain both DNA replication timing precision and 3D genome organization integrity. Cell Rep. 2021;36(12):109722. doi:10.1016/j.celrep.2021.109722
DNA 复制时间和三维 (3D) 基因组组织与 large domains 的不同表观基因组模式相关。然而,表观基因组的改变,特别是与癌症相关的 DNA 低甲基化,是否会影响更高层次的基因组结构仍不清楚。在这里,我们使用 Repli-Seq、单细胞 Repli-Seq 和 Hi-C,表明全基因组甲基化丢失与复制时间精度的一致丢失和 3D 基因组组织的失调有关。值得注意的是,我们发现 3D 基因组划分明显中断,细胞间复制时间异质性的显着增加和癌症低甲基化模型中等位基因复制时间的丢失,可能是通过 DNA 复制和基因组组织途径的基因失调。最后,我们从大型低甲基化域中识别出异位 H3K4me3-H3K9me3 域,在这些域中保持晚期复制,我们声称这有助于防止灾难性的基因组重组和异常基因转录。我们的结果强调了甲基化组在维持 3D 基因组调控中的潜在作用。
我的理解
DNA甲基化在DNA复制和维持基因组结构的稳定性发挥非常重要的作用,但是作者使用的DNA甲基化测序技术没接触过。
- 利用汇总统计imputation在小样本情况下加强分子QTL研究的发现
Enhancing discoveries of molecular QTL studies with small sample size using summary statistic imputation
https://pubmed.ncbi.nlm.nih.gov/34545927/
Wang T, Liu Y, Yin Q, et al. Enhancing discoveries of molecular QTL studies with small sample size using summary statistic imputation [published online ahead of print, 2021 Sep 20]. Brief Bioinform. 2021;bbab370. doi:10.1093/bib/bbab370
多组学分子特征的定量特征位点(Quantitative trait locus, QTL)分析,如基因转录(eQTL)、DNA甲基化(mQTL)和组蛋白修饰(haQTL),已被广泛用于推断基因组变异的功能效应。然而,QTL的发现在很大程度上受到有限的研究样本量的限制,这要求小等位基因频率的阈值更高,然后导致大量缺失分子特征变异关联。由于样品的可用性和成本,这在单细胞水平分子QTL研究中非常明显。迫切需要提出一种解决这个问题的方法,以加强目前样本量小的分子QTL研究的发现。在这项研究中,我们提出了一个有效的计算框架,称为xQTLImp,以将缺失的分子QTL关联进行计算。在局部区域指责中,xQTLImp 使用多变量高斯模型,利用已知的变体关联统计数据和周围联系不平衡 (LD) 来将缺失的关联进行推定。在全基因组的计算中,采用了新的程序来提高效率,包括动态构建重复使用的LD缓冲器,采用多种启发式策略和并行计算。对各种多omic散装和基于单细胞测序的QTL数据集的实验表明,xQTLImp具有较高的计算精度和新型QTL发现能力。最后,https://github.com/stormlovetao/QTLIMP 可免费获得C++软件包。
关键词: QTL分析;插补框架;单细胞;样本量小;汇总统计。
- 多组学分析揭示多灶性肝内胆管癌全面的肿瘤异质性和不同的免疫亚型

目的: 靶向治疗和免疫治疗正在改变肝内胆管癌(ICC)的治疗方法。然而,关于多灶性 ICC 的肿瘤间异质性 (ITH) 及其对患者对这些治疗反应的影响知之甚少。我们旨在表征多灶性 ICC 的免疫基因组和表观基因组异质性,以指导治疗决策。
实验设计: 我们从 16 名多灶性 ICC 患者中获取了 66 个肿瘤样本,并使用全外显子组测序、群体和单细胞 RNA 测序、甲基化微阵列和多重免疫染色来表征肿瘤和免疫异质性。根据中位 ITH 指数将患者分为高或低 ITH 组。两个独立的队列被用来验证发现。评估了对抗 PD-1 治疗的反应。
结果: 多灶性 ICC 在高 ITH 组患者中表现出相当大的肿瘤间基因组、转录和表观基因组异质性。在高或低 ITH 组中,患者体内多个肿瘤的免疫谱相对较少异质,并且观察到多个肿瘤对抗 PD-1 免疫治疗的一致反应。免疫标志物的无监督聚类确定了一种低免疫亚型和一种高免疫亚型,具有更高的免疫细胞浸润、更密切的肿瘤-免疫细胞相互作用和高免疫亚型中的IFN特征表达上调。确定CD8B和ICOS 的表达水平促进了这种免疫分类和患者预后的预测。最后,启动子 DNA 甲基化通过调节免疫基因表达导致两种亚型的不同免疫谱。
结论: 多灶性ICC的基因组、转录组和表观基因组存在全面的异质性。基于ICC的异质性较低的免疫特征,我们建议对患者的预后进行分层并可能支持个性化免疫治疗的免疫分类。
- DNA 甲基化和 microRNA 表达的综合分析揭示了肝细胞癌中种族异质性的机制

背景:表观遗传调控的病理改变长期以来被认为是许多癌症的标志,包括肝细胞癌 (HCC)。在健康个体中,DNA 甲基化与 microRNA (miRNA) 表达之间的关系保持着良好的平衡;然而,这种和谐的破坏可能有助于癌症的发生或现有癌症的传播。DNA 甲基化和 microRNA 表达之间的平衡及其在 HCC 中的潜在干扰可能因种族而异。有新的证据将表观遗传事件(包括 DNA 甲基化和 miRNA 表达)与癌症差异联系起来。
实验设计:在本文中,我们通过对 DNA 甲基化、miRNA 和基因表达联合调控的综合分析来评估 HCC 中人群异质性的表观遗传机制。具体来说,通过对患有 HCC 的非裔美国人 (AA) 和欧洲裔美国人 (EA) 的肿瘤和邻近非肿瘤肝组织进行分析,获得DNA 甲基化, mRNA-seq 和 miRNA-seq 数据。
结果:使用混合方差分析,我们确定了胞嘧啶-磷酸-鸟嘌呤 (CpG) 位点、mRNA 和 miRNA,这些位点、mRNA 和 miRNA 在 HCC 与相邻非肿瘤组织中以种族特异性方式显着改变。我们观察到 EA 中的甲基化组发生了巨大变化,其差异甲基化和差异表达的基因数量明显多于 AA。另一方面,与 EA 相比,AA 中 miRNA 表达的改变程度更大。通路分析在功能上将 EA 中的表观遗传调控与涉及免疫细胞成熟、炎症和血管重塑的过程联系起来。相反,细胞增殖、新陈代谢、作为这种表观遗传分析的结果,发现生长途径在 AA 中占主导地位。此外,通过综合分析,我们确定了具有不同表观遗传调控的 HCC 中显着差异表达的基因,这些基因与 AA 的 miRNA 表达变化和 EA 的 DNA 甲基化有关。
- scMethBank:单细胞全基因组 DNA 甲基化图谱数据库
数据库访问地址:https://ngdc.cncb.ac.cn/methbank/scm/
单细胞亚硫酸氢盐测序方法被广泛用于评估细胞状态的表观基因组异质性。在过去几年中,产生了大量数据并促进了对许多关键生物过程的表观遗传调控的更深入理解,包括早期胚胎发育、细胞分化和肿瘤进展。构建海量数据的功能性资源平台迫在眉睫。在这里,我们展示了 scMethBank,这是第一个开放访问的综合数据库,专门用于单细胞 DNA 甲基化数据和元数据的收集、整合、分析和可视化。scMethBank 的当前版本包括处理过的单细胞亚硫酸氢盐测序数据和来自 15 个公共单细胞数据集的 8328 个样本的精选元数据,涉及两个物种(人和小鼠)、29 种细胞类型和两种疾病。总之,scMethBank 旨在通过提供浏览、搜索、可视化、下载功能和用户友好的在线工具,帮助对细胞异质性感兴趣的研究人员在单细胞水平上探索和利用全基因组甲基化数据。

若有收获,就点个赞吧
0 人点赞
