简介
cfDNA是疾病液体活检的一种强大工具,而cfDNA的表观遗传修饰对于疾病检测和分类也是至关重要的。
作者使用公共数据库数据,开发了标准化质控、分析流程,并构建了一个网络服务器,以在基因组规模上查询、可视化5mC、5hmC和核小体定位信息。
使用该数据库,用户能够:
1,浏览来自血浆或血清的cfDNA的表观遗传信息
2,浏览癌症发展不同阶段(早期、晚期或转移期)的cfDNA表观基因组
3,USCS基因组可视化和比较
4,下载数据
5,使用CFEA流程分析公共或私有cfDNA表观基因组数据。
数据库发表
CFEA: a cell-free epigenome atlas in human diseases
获取地址:https://academic.oup.com/nar/article/48/D1/D40/5552060
代码获取地址Github:https://github.com/lemonsky123/CFEA-pipeline(推荐)
发表时间:20 August 2019;Nucleic Acids Research
摘要:表观遗传变化,包括5-methylcytosine (5mC), 5-hydroxymethylcytosine (5hmC) and nucleosome positioning (NP),在Cell-Free DNA(cfDNA)中被广泛观察到,许多可用的基于cfDNA的表观基因组全型剖面在疾病检测和分类中表现出高度的敏感性和特异性。然而,由于缺乏有效的收集、标准化的质量控制和分析程序,有效整合和重用这些数据仍然是相当大的挑战。在这里,我们介绍了CFA(http://www.bio-data.cn/CFEA),一个Cell-Free表观基因组数据库,专门用于三种广泛采用的表观遗传修饰(5mC,5hmC和NP)涉及27种人类疾病。我们开发了用于质量控制和标准数据处理的生物信息管道以及易于使用的 Web 界面,以方便查询、可视化和下载这些无细胞表观基因组数据。我们还手动为每个配置文件精心策划相关的生物和临床信息,使用户能够更好地浏览和比较癌症发展的特定阶段(如早期或转移阶段)的 cfDNA 表观基因组。CFEA为科学界提供了全面和及时的资源,并支持开发用于各种人类疾病的液体活检生物标志物。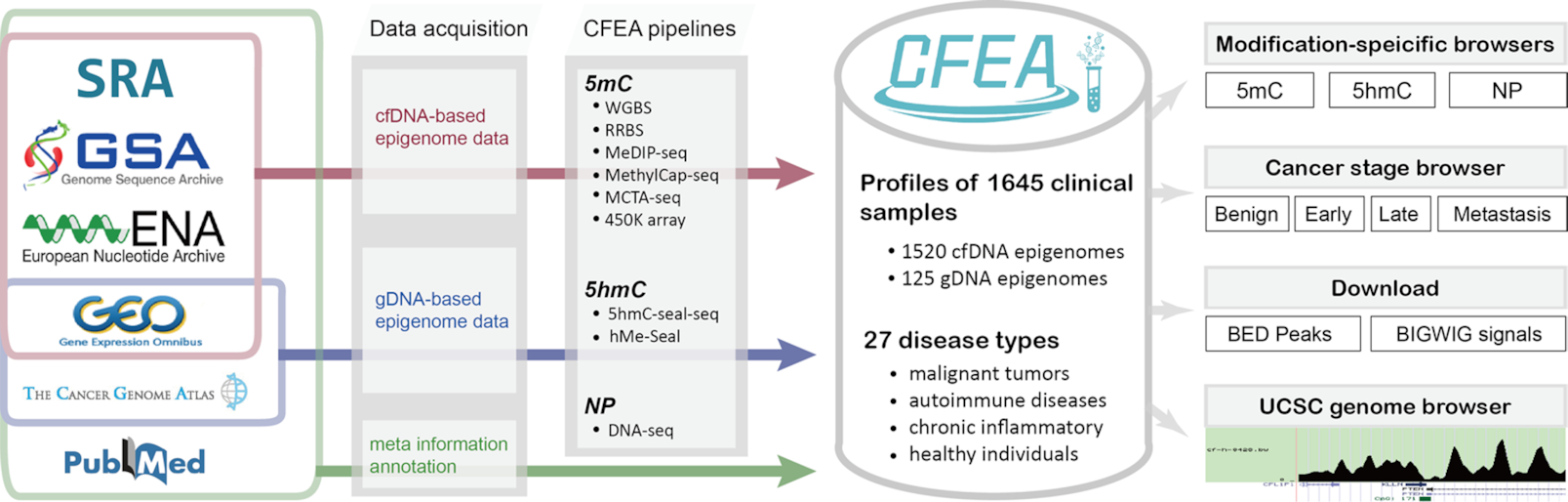
图1 | CFEA数据库的示意图
CFEA 数据库主页:http://www.bio-data.cn/CFEA/index.jsp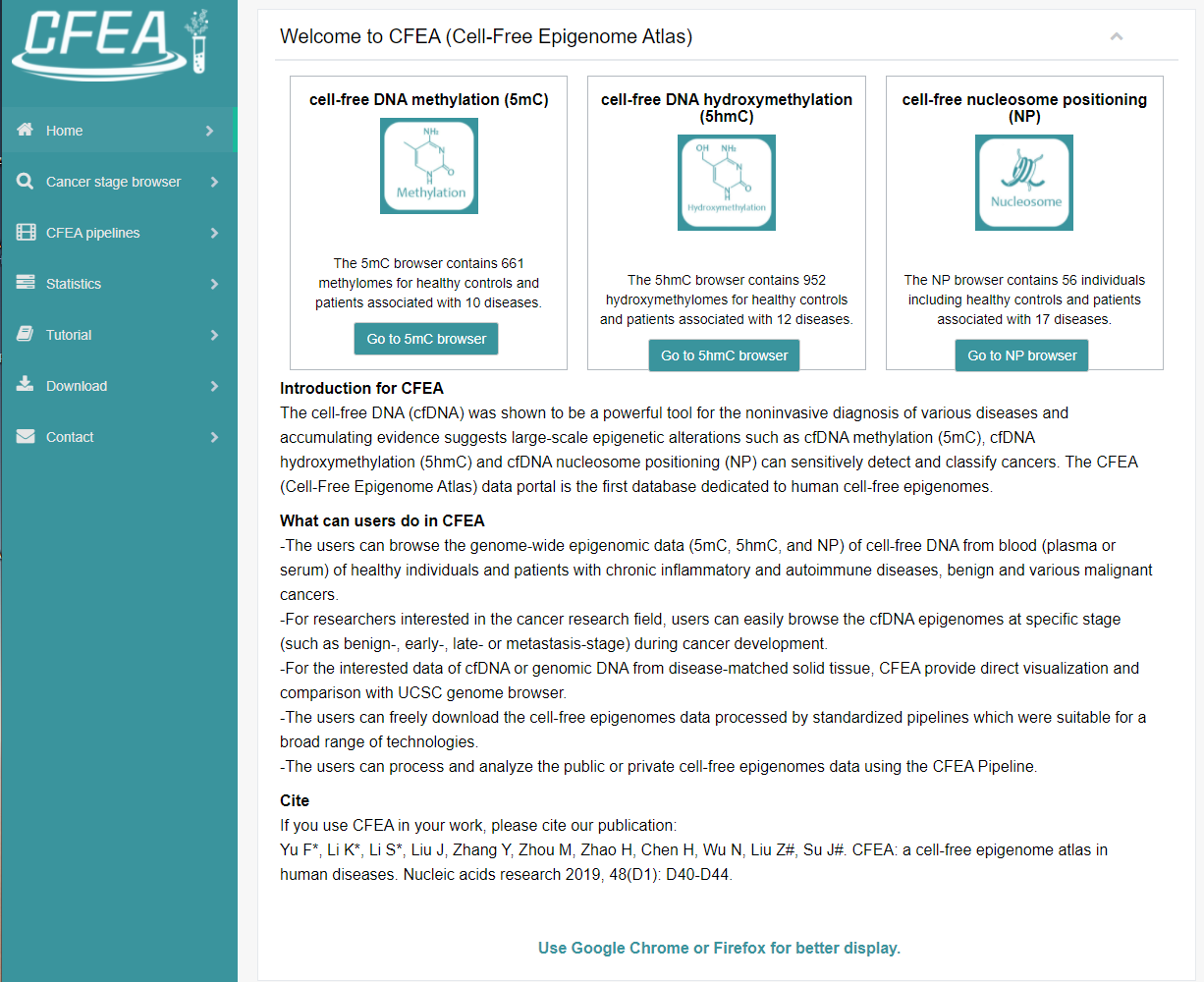
Details about CFEA pipeline
CFEA的分析流程介绍,目录如下:
- Overview of Pipelines
- Software Requirements
- Quality controls
- WGBS/RRBS (5mC)
- Seal-seq/hMe-Seal-seq data (5hmC) & Methyl-Cap/MeDIP-seq data (5mC)
- MCTA-seq data (5mC)
- 450K data (5mC)
- DNA-seq (NP)
Overview of Pipelines
除cMethDNA检测(低通量DNA甲基化检测实验)外,所有检测技术(9种)均由我们的管道覆盖,我们直接从GEO网页下载beta值为基础的配置文件。用法的详细描述如下所示: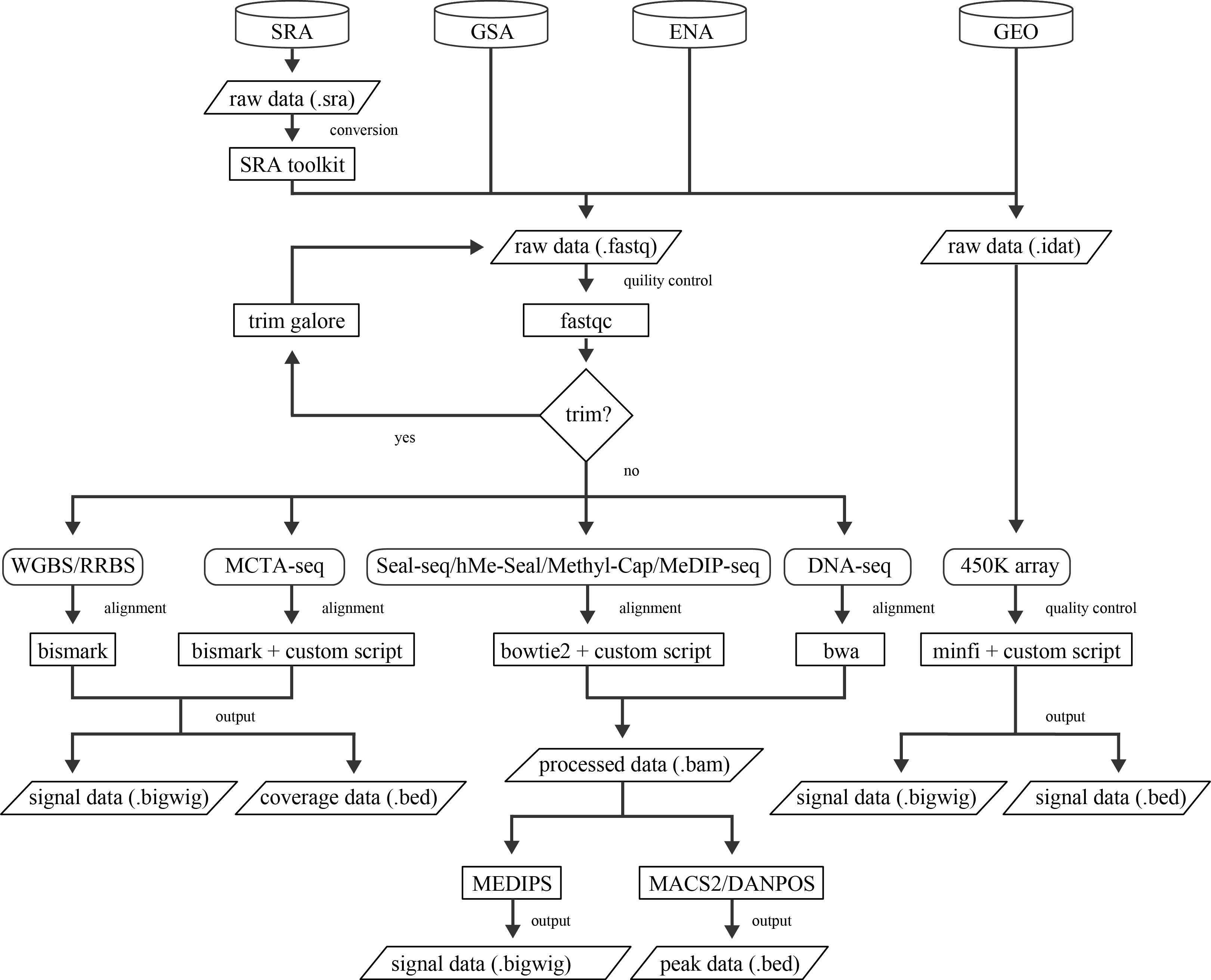
Software Requirements
We recommend you to install all the softwares into a local environment before starting the data analysis. Conda may be a good option for this purpose.
conda create -n CFEA python=2.7conda install -n CFEA fastqc bismark bowtie2macs2 picard multiqc samtools deeptools
代码获取地址:https://github.com/lemonsky123/CFEA-pipeline
Quality controls
我们想在比对前检查一下测序数据的质量。使用fastqc fastq.py脚本,我们使用fastqc软件评估原始fastq序列的质量,并使用MultiQC软件将fastqc结果收集到一个报告文件中。
python raw_fastq_qc.py -i [path to you fastq files] -p 10 -q [qc result dir]-l [directory to log files] -n [project name in MultiQC]
在完成上面的清理步骤之后,我们将介绍如何为来自不同技术的不同数据类型使用我们的管道。
WGBS/RRBS (5mC)
全基因组亚硫酸氢盐测序(WGBS)和简化代表性亚硫酸氢盐测序(RRBS)都能在单碱基分辨率下得到亚硫酸氢盐转换的测序数据,因此可以使用相同的处理程序。对于修剪步骤,我们使用工具trim_galore。修剪后的数据然后让bismark比对到参考基因组,命令如下。
trim_galore --rrbs --illumina --phred33 --paired [fastq_file_1][fastq_file_2] -o [clean_out_dir] > [log_file] 2>&1bismark [bismark_index] --path_to_bowtie [bowtie2_path] --bowtie2 -1[clean_file_1] -2 [clean_file_2] -o [this_sample_mapping_out]--temp_dir [temp_dir] > [this_sample_bismark_log] 2>&1
包装后的python脚本(mapping_WGBS.py)的命令如下。因为WGBS需要去除PCR重复,而RRBS不需要。
python mapping_bismark.py -i [path fastq] -p [numer of processes] -m[mapping result dir] -l [log dir] -b [path to put bam files]-n [project name for multiQC] -t [bowtie2 path] -d [bismark index]-c [path to put trimmed fastq files]
在上述步骤之后,我们将得到对齐的bam文件。接下来,我们将使用bismark甲基化提取器从对齐的bam文件中提取甲基化覆盖范围。
bismark_methylation_extractor -s [bam] --bedGraph --counts -o[this_cov_dir] > [this_log_dir] 2>&1
下面使用运行此命令的python脚本(extract_methylation_coverage.py)。
python extract_methylation_coverage.py -i [bam dir] -p [number of processed]-c [coverage out dir] -l [log dir]
bismark_methylation_extractor的输出包含正链和负链CpGs,所以我们使用下面的脚本来合并这些位置。【之前我也想过自己写脚本合并,发现还是找轮子比较方便】
python merge_cpg_pos.py -i [coverage dir] -p [number of processes]-f [whole genome CpGs]
在执行上述步骤之前,必须首先使用脚本获取整个基因组cpg(get_cpg_whole_genome.pl)。
perl get_cpg_whole_genome.pl hg19.fa [whole genome CpGs]
最后,要将coverage格式文件转换为wiggle格式。
python coverage_to_wig.py [coverage dir]

若有收获,就点个赞吧
0 人点赞
