本文根据 2016 年 8 月北京大学肿瘤医院邓大君教授在「表观基因组学暑期国际讲习班」中的报告整理而成,本文采用第一人称叙述,文中的 “我” 皆指邓大君教授。报告原视频详见:系列视频 4 | 北大肿瘤医院邓大君:表观基因组学与肿瘤发生发展和转移,视频全长约 3h22min,本文约 2 万字。
邓大君,北京大学肿瘤医院病因学研究室主任,教授,研究员,博士生导师;1986 年中国协和医科大学研究生毕业,1992、1994、1996 年德国联邦药物所毒理系客座科学家,2001 年 5 月至 2002 年 4 月在美国弗吉尼亚大学医学中心做高级访问学者;享受国务院政府特殊津贴,北京市科技新星,新世纪百千万人才工程国家级人选。
肿瘤表观遗传学领域每天有大量文章产生,不断刷新我们的观念和认知。本文仅代表 2016 年 8 月邓大君教授的观点。总的来说,本文信息量很大,邓教授的很多观点非常具有启发性,建议收藏慢慢消化。
以下为正文:
我从事了三十年的肿瘤研究,很高兴今天在这里跟大家切磋一下什么是肿瘤,什么是肿瘤的表观遗传。在座的有很多不是医学背景的,因此我讲解的时候会适当的做一些科普,照顾一下非医学背景的同学;但同时还要兼顾在研究第一线的老师,希望让多数人能在这三小时里受益。
在开始讲之前,我想做一个简单的科普——肿瘤是什么,然后再说一下肿瘤发生发展的特征,在这基础之上,咱们再来谈论表观遗传的本质和肿瘤发生发展的关系,以及除了它们科学意义以外,还能用它来干什么,比如在肿瘤诊断治疗上有些什么具体的用途。我们都知道肿瘤已经被研究了上百年,表观遗传也已经火了二十多年,已经到了可以解决一些问题的时候。
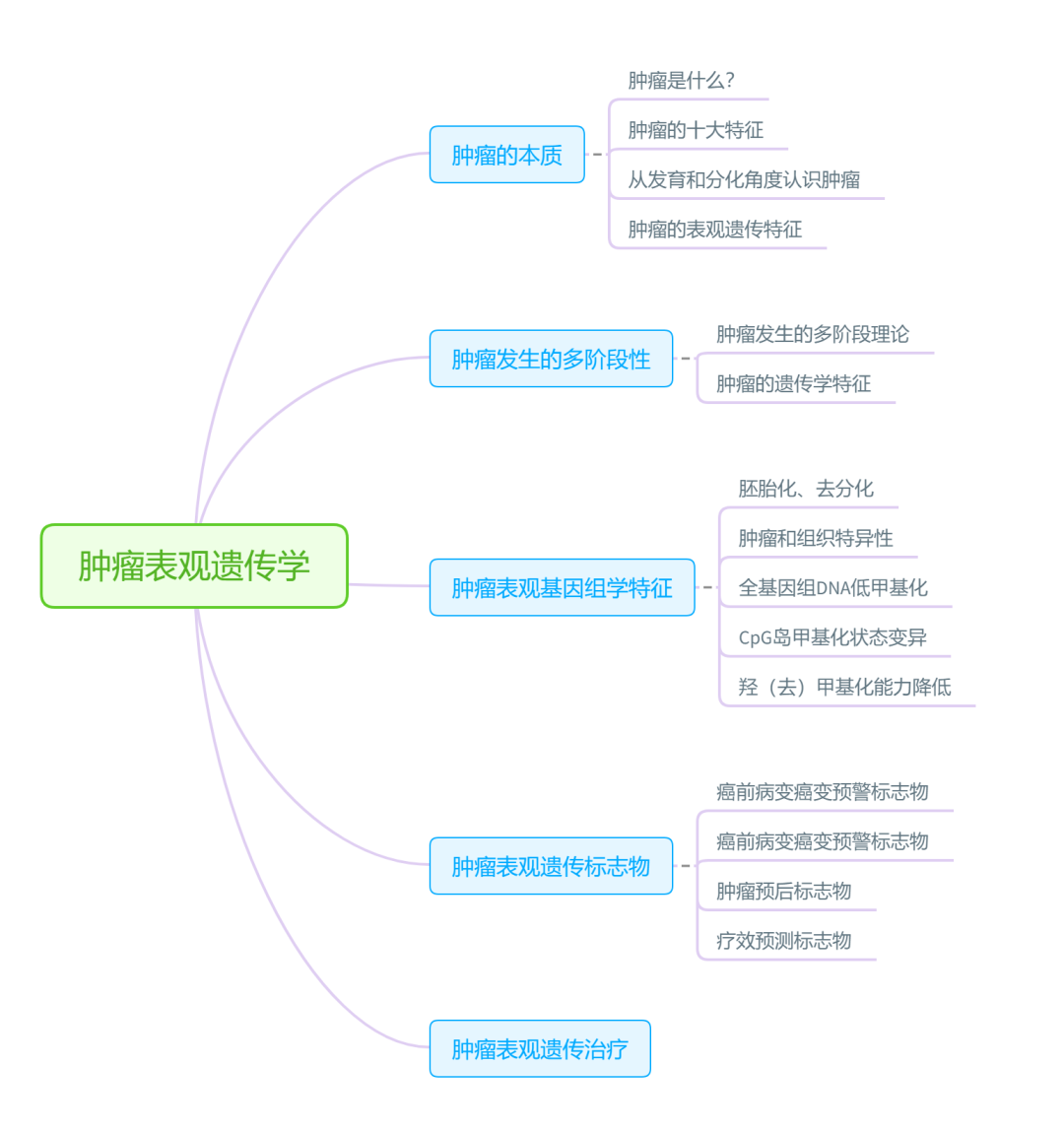
本文的思维导图
肿瘤的本质
肿瘤是什么?
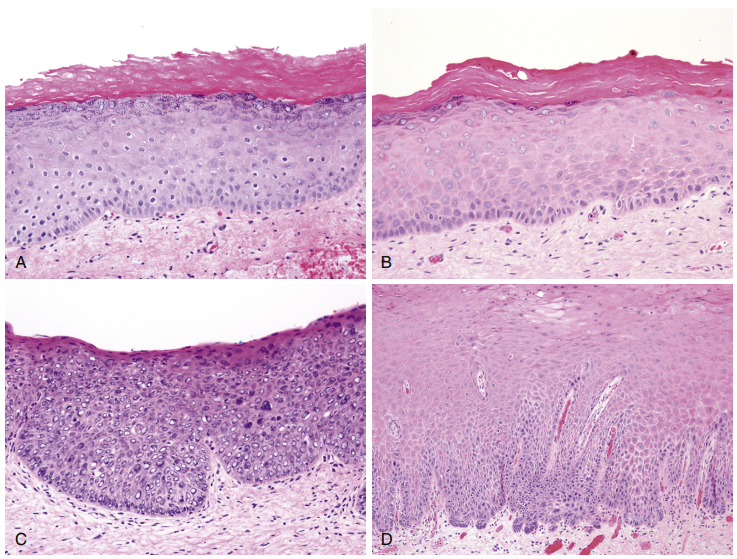
图 1. 鳞状异常增生。A. 良性的角化病;B. 轻度鳞状异常增生;C. 重度鳞状异常增生;D. 鳞癌。[1]
这是鳞状上皮的结构,图 1A 中下面是结缔组织,中间是基底膜,上面是基底层的鳞状上皮,上面是角化层。口腔黏膜的角化层相对比较薄,我们头皮的角化层会比较厚,上面基本看不见细胞核,角化细胞是没有核的。正常组织是一层一层的,层次分明;而在图 1D 中可以看到上皮细胞异常增生,穿透基底膜到结缔组织里,这就是所谓的肿瘤,它有结构的异形性,也有细胞的异形性。
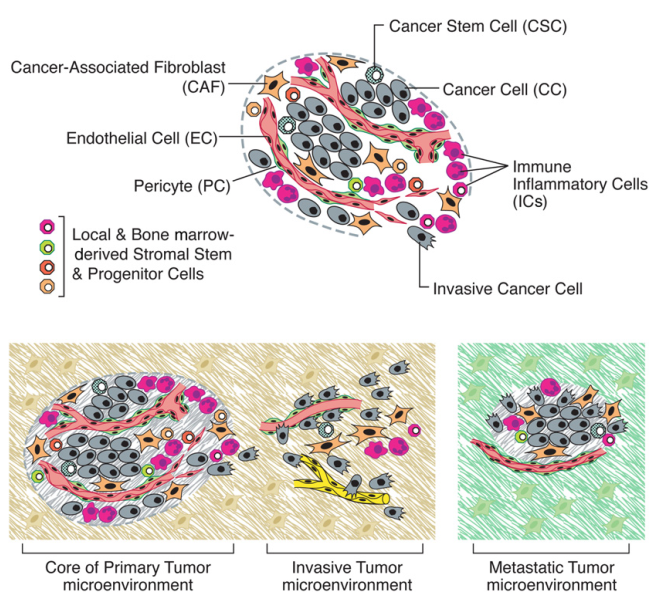
图 2. 肿瘤微环境 [2]
这些 HE 染色的片子给了我们关于肿瘤的基本印象,如果从细胞生物学的角度,实际上肿瘤组织里面并不是每一个都是肿瘤细胞,其中还有我们正常机体的结缔组织。肿瘤组织里面包括肿瘤细胞,边上有结缔组织,还有炎症细胞、纤维细胞等。当正常组织里面出现了一个肿瘤细胞时,这个肿瘤细胞会有什么作用呢?就像我今天站在这里对你们宣传和推销,这个肿瘤细胞可能把周围所有的细胞给同化了。虽然它们不是变成了肿瘤细胞,但它们变成了 “帮凶”,所以这是 “宣传” 的力量。这也就是所谓 “种子” 和“土壤”的概念。肿瘤细胞有很多同化的手段,比如外泌体,可以把它分泌的东西导到别的细胞上去。
肿瘤的十大特征
肿瘤有六大基本特征,无限增殖潜能、侵袭和转移性、失去接触抑制、自主增殖信号、对抗程序性死亡、诱导血管形成。肿瘤细胞不仅能够自主增殖,还有侵袭性。恶性肿瘤会把周边的正常组织给破坏了;而良性肿瘤可以无限增殖,但是它不破坏,可以长得很大。一个良性的卵巢肿瘤,可能长到上百斤,但它不会要了你的命。但如果它会侵袭破坏周围的正常组织,往往意味着生命就将不幸终结。恶性肿瘤不仅破坏周围的正常组织,还可以通过血液循环和淋巴道扩散到远处,这就是所谓的侵袭转移能力。
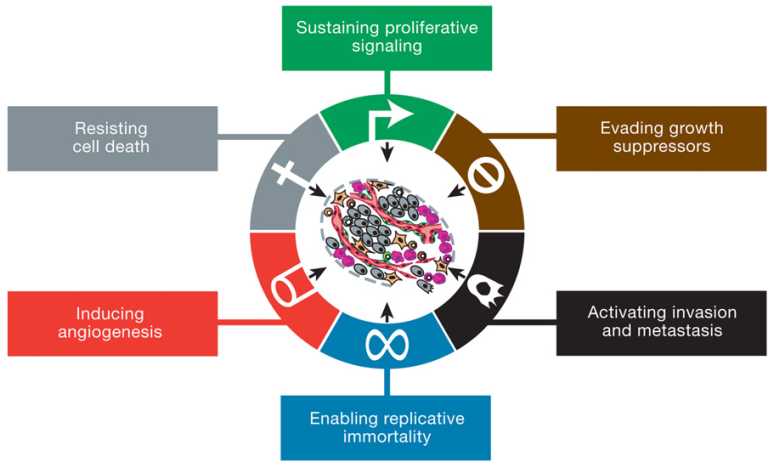
图 3. 肿瘤基本的六大特征 [2]
组织培养体系建立以后,人们发现把肿瘤细胞拿出来以后,它的生长状态跟正常细胞有差异。正常细胞从组织中取出原代培养是有规律的:往外长,但两个纤维细胞相互接触后,会产生接触抑制,就不长了。但肿瘤细胞不是这样,失去接触抑制,它会摞起来长。无限增殖潜能、侵袭和转移性和失去接触抑制是可以从形态学上观察到的最主要的特征。
随着对肿瘤分子生物学认知的提高,开始加入了新的特征,比如自主增殖信号。为什么肿瘤细胞有无限增殖能力呢?我们的很多细胞自己独自是生长不了的,它需要生长激素。肿瘤细胞可通过自分泌或旁分泌的模式来满足自我生长,而不依赖其他细胞。上世纪 80 年代后期凋亡现象发现以后,发现肿瘤细胞不一定长的快,但它不死,最后肿瘤也变大。肿瘤还可以诱导血管生成。这是对肿瘤早期的认识。
近些年又发现了肿瘤的几个新特征:能量代谢异常、基因组不稳定、避免免疫破坏、诱发炎症反应。
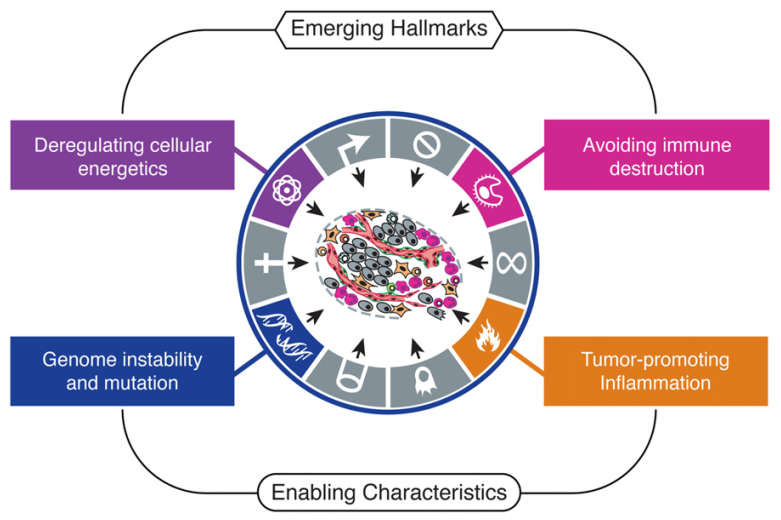
图 4. 肿瘤的新的特征 [2]
关于肿瘤的能量代谢异常,大家肯定听说过 “Warburg 效应”。最初很多人以为是因为肿瘤长大了,血泵不够,所以缺氧,因此选择糖酵解途径。但实际上肿瘤在很小的时候,不需要额外血管的时候,肿瘤细胞就呈现糖酵解的特征,为什么?虽然经典的氧化磷酸化途径会生成很多的 ATP,但效率很低;糖酵解效率很高,而且同时生成很多肿瘤细胞蛋白质合成、核酸合成需要的底物。后来发现肿瘤的一个很显著的特征是基因组不稳定,这个是肿瘤发生的早期现象。肿瘤的免疫逃逸大家应该听了很多。正常情况下,肿瘤细胞会形成一种免疫抑制,可以在人体内活下来。肿瘤的促炎症反应也是一个很重要的特征。我们今天要讲的表观遗传的问题,跟这十大特征密切相关。
从发育和分化角度认识肿瘤
受精卵经历复杂的分化发育过程,形成了拥有 200 多种不同类型细胞的我们。在显微镜下观察,我们发现肿瘤细胞好像有点返祖了,出现了去分化、胚胎化。很多在胚胎时期表达的基因,在肿瘤组织里重新表达。这是不是意味着在我们的组织中,已经完成分化的终末分化细胞又重新去分化了,变成了分化初始状态的细胞?
早期没有干细胞概念的时候,以为是肿瘤细胞发生了去分化,直到后来我们有了肿瘤干细胞的概念。实际上现在基本的观念认为,每个组织中都有正常的组织干细胞(tissue stem cell),这些细胞平时进行非常轻微的分裂活动。我们的组织需要更新,在更新的时候起到了细胞起源的作用。比如我们的胃肠道细胞,一星期全部更换一次;而我们每天都会掉头发,但很快又重新长出来;造血细胞每天都重新生成新的白细胞、红细胞。这就是组织干细胞存在的意义
组织干细胞本身它还是要服从组织的控制,不该分裂时不分裂,但当它变成肿瘤以后,就不受管控。这时候这种组织干细胞的分化状态就不是终末分化了。当它占比增高的时候,显微镜下观察会认为这个肿瘤组织有点返祖或去分化了,实际上并不是去分化,而是原始状态细胞的比例越来越多。而且我们的机体里大部分的肿瘤来自于组织干细胞。即使是正常的干细胞,如果没有一个合适的环境,它可能会长成肿瘤。反过来,即使是肿瘤细胞的基因组,把它放到一个合适的环境里,它会发育成正常的机体。
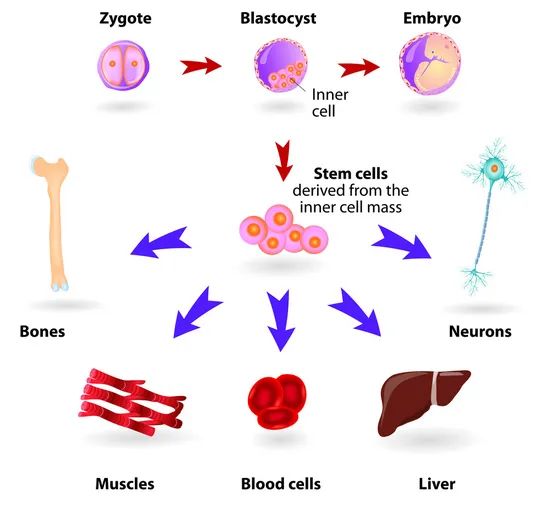
图 5. 胚胎干细胞具有全能性
图源:https://www.medicalsciencenavigator.com/stem-cells-an-evolving-definition/
胚胎干细胞具有全能性,它卵裂成八个细胞后,如果把这八个细胞分离,它们可以成为八个独立的胚胎。我们都听说过双胞胎,人类的极限就是八胞胎,没有更多了。胚胎干细胞有无限增殖能力,曾经有人试图利用它的无限增殖能力,来把这个细胞种到人体组织 “坏掉” 的地方,因为它可以体外培育。种到体内之后可以诱导它分化,生成正常的组织器官。比如经常喝酒的人肝坏了,经常抽烟的人肺坏了,一个设想是在这些地方放入干细胞。肝和肺的组织结构复杂,但有一些器官或组织结构不复杂。比如帕金森综合症病人分泌多巴胺的神经元减少了,把胚胎干细胞诱导成能分泌多巴胺的细胞,种到患者大脑中神经元所在的部位,那是不是就可以治疗帕金森氏综合症?这个事在 2002 年的时候的的确确发生过,当时干细胞注射到病人趋化多巴胺神经元那个部位去[3],短期内病人手不再颤抖了,但没过两年,长成肿瘤了[4]。这些研究提示,生长不可控不仅是肿瘤的特征,正常的干细胞如果没有合适的环境,也会生长不可控。现在国家推动了很多 iPS 细胞(induced pluripotent stem cells,诱导性多能干细胞)的项目,用 iPS 细胞做基因治疗,仍然面对的一个问题——可能会长出肿瘤。
唯一成功的就是脐带血干细胞,因为它已经完成了一定的分化。现在很多人用它来治很多病(批准的不批准的……),还有人用它来保养、防衰老、祛皱,就像万金油一样,什么都给你打两针脐带血干细胞,但到底哪些病能治哪些病不能治还不知道。不过确确实实,有些病通过注射脐带血干细胞是可以治疗的。脐带血干细胞它的循环特征是血液里,注射后细胞可以跑到全身去,它对环境的要求就和其它的干细胞是不一样的。在干细胞治疗上,脐带血干细胞是比较成功的。
肿瘤细胞不是我们想象的那样凶形恶煞。家庭里出了坏孩子,他还是孩子,没有本质区别。正常细胞和肿瘤细胞之间没有那么大的区别,你给它一个正常环境,它就可以浪子回头。如果我们可以开发出这样一种肿瘤的治疗方法那该多好。
小结一下:正常细胞在正常环境下才能正常生长;肿瘤细胞在正常环境下可能正常生长,肿瘤细胞在肿瘤环境会生长成肿瘤,但正常细胞在肿瘤的环境下也会长成肿瘤。
肿瘤的表观遗传特征
我们刚才提到了受精卵最后分化成 200 多种不同的细胞,而细胞分化过程是主要靠表观遗传决定。分化相关的表观遗传非常稳定,有多稳定呢?皮肤被刀划破了,长回去的还是皮肤;骨头摔折了,长回去的还是骨头。它牢牢的记得自己是干什么的,来自哪里。哪怕把细胞从体内切下来放到培养瓶培育,它还长得像原来的样子,不会变成别的细胞,除非你用类似 iPS 细胞的那种模式去干扰它的基因。
与此同时,肿瘤的发生也暴露在一个环境中,所以它还是表现出一定的适应于环境的变化,而环境适应性表观遗传是相对不稳定的。机体除了稳定地维持 200 多种细胞存在以外,还要生长在不同的环境中,无论是西藏海拔四、五千米的地方,还是北极极寒的环境,我们都可以依然生存,机体形成了强大的适应能力。当然,营养的好坏也有影响。我们这一辈五六十年代出生的人,没吃没喝,所以形成了一种所谓的 “节约型代谢模式”;像你们有吃不完喝不完的,是 “富裕型代谢模式”。而且这种模式在你们的基因组里面牢牢的记住,不仅是自己这一代,还会传递给下一代。这就是表观遗传,不仅是适应,还可以传递下去。这也就是为什么说表观遗传很重要的原因。
关于肿瘤的本质就介绍到这里,希望大家对它有个清醒的认识,别把它理解成敌人,“坏孩子” 也是咱们自己家的。
肿瘤发生的多阶段性
肿瘤发生的多阶段理论
罗马不是一天建成的,肿瘤也不是一下子就变成肿瘤了。机体大部分正常组织要变成肿瘤,不是那么容易,家族性的例外。如果某一个很重要的基因在身体的每个细胞中已经失活了,机体如果还有另一次打击(二次打击学说[5]),细胞就可能要癌变了。我们在座的长到这个年纪没得肿瘤,遗传性的肿瘤基本上不会有;而对于非遗传性(散发性)肿瘤,多数人得在 55 岁以后才会出现,如果 70、80 岁还没有得,那就 “老不死”。
肿瘤的发生是一个漫长的过程。肿瘤的发生可以分成启动阶段、促进阶段和进展阶段(演变阶段)。在启动阶段,不管是化学致癌物、物理致癌物,还是生物致癌物作用于正常细胞后,细胞变得有 “不听话” 的潜能。但它不是真正的肿瘤,不是正常的细胞了。它就是个单一细胞,不像肿瘤那样是一大块。不管它恶性的程度有多高,在我们每个人体内都有,而且还很多,但绝大多数没有机会长成肿瘤。因为它跟边上所有的细胞建立了联系,被内分泌系统等很多因素控制住,成不了气候。但如果这个细胞的生存环境出现了致癌物,使它不听话;如果一些因素把细胞之间紧密连接的 gap junction 切断,这个细胞就和周围组织环境失去了接触,就会生长起来,我们称之为“克隆性增殖”,成为一种潜在的病灶。随着我们诊断手段的提高,可以发现的克隆越来越小,可能是一毫米、半毫米。尤其是在显微镜下发现一群异常细胞,由于还比较小,不会要你的命。只有恶性程度越来越高的细胞,不断演变并进入演变阶段,进一步变成早期肿瘤、晚期肿瘤、侵袭性肿瘤。
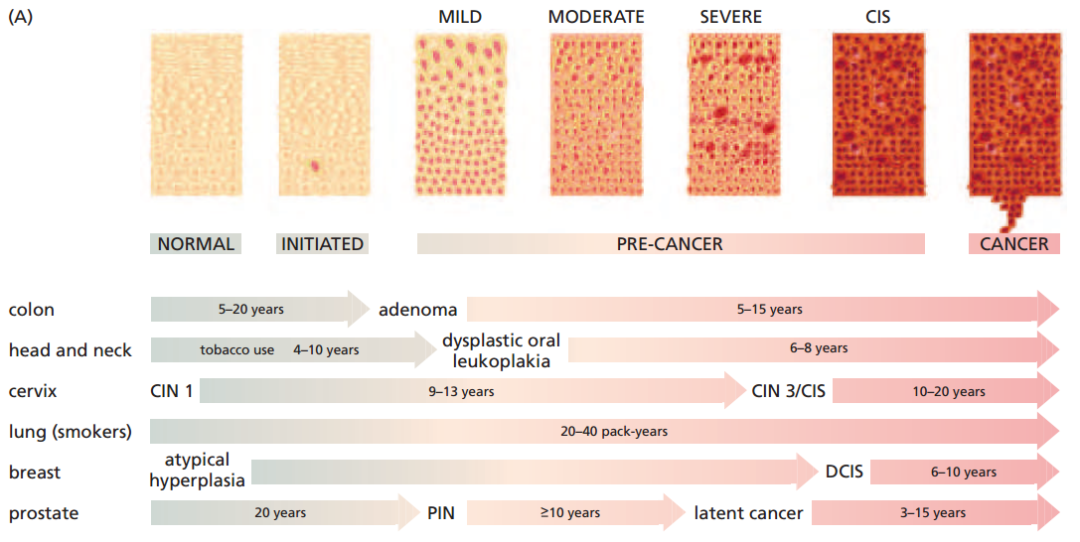
图 6. 不同器官部位的多步骤肿瘤发生过程 [6]
针对不同的阶段需要采取不同的策略。在启动阶段(正常细胞),如果不接触致癌物基本就不会得肿瘤。如果我们接触的东西没有带致癌的,它可能没有机会增殖起来(促进阶段)。如果到了癌前病灶阶段,其实只有少数的癌前病灶会长成肿瘤。我们会建立一系列的标准来识别它,谁会癌变谁不会癌变,这是肿瘤研究中非常重要的一块。而一旦到了临床前恶性肿瘤阶段,我们就要进行筛查。很多人不愿意去医院,因为有些检查不舒服,比如肠镜、内窥镜,所以你得找到理由让人们去做,一般是通过筛查看有没有某个指标高。到了临床期恶性肿瘤阶段,如果跑了周围,我们可以切掉;如果跑到全身了,就切不掉,只能用放疗、化疗、免疫治疗。但到底用什么治疗,用什么药呢?要有一个疗效预测的标准。真要是转移到远处去(晚期恶性肿瘤阶段),那基本就是神仙也救不过来了。少数肿瘤例外,如果是单一转移灶,还可以切掉,病人活的可能更长一下,治好是不太可能的。肿瘤如果在临床前和临床期阶段发现了,还是有可能治好的。所以肿瘤发展的多阶段性决定了我们做肿瘤表观遗传的时候明确任务是什么。很多人不明白,做肿瘤要做什么事,就看你定位在哪个阶段,所以不同的阶段,功能和性质是不一样的。
肿瘤的遗传学特征
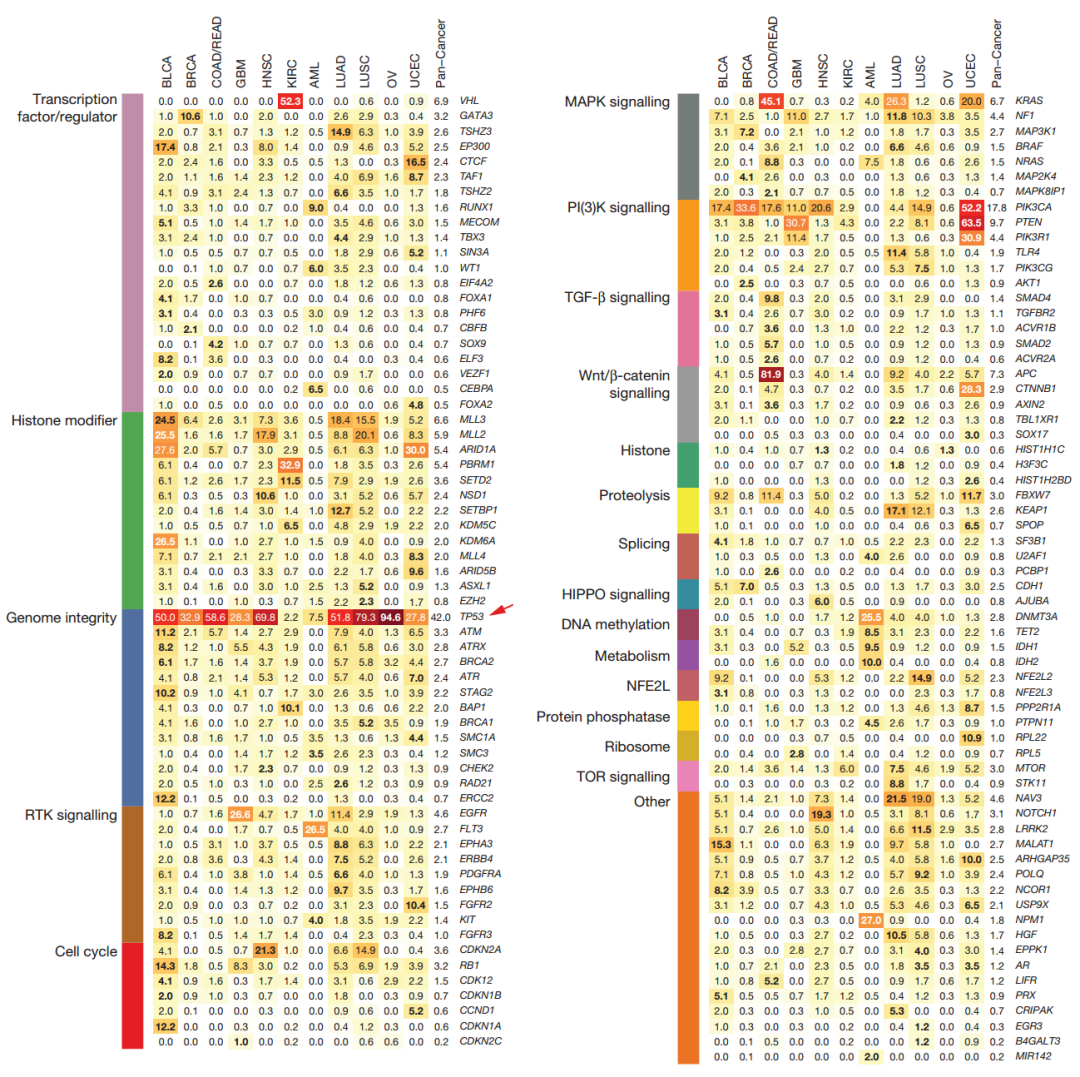
图 7. 人体 12 种恶性肿瘤组织中常见基因突变百分率 [7]
全世界已经测了大量肿瘤的基因组了,测出了很多突变。图 7 中,颜色深的地方表示基因突变百分率就高,其中 TP53 基因是最明显的。当然这个是指突变,基因的变化有很多,比如拷贝数变异、deletion、insertion,还有很多变化现在还测不了。我们测出来了这么多的变化,但还是有个问题。我们做肿瘤的有时候会很迷惑,在肿瘤患者中测出来一个突变,发现有突变的人,比没突变的人活得好。我们不理解,为什么有突变的比没突变的活得好;另外,我们还不知道那个没有突变的肿瘤是怎么发生的,这提示当前的研究还有一块很重要的东西没把握到。
实际上,回过头来看,很可能是基因组不稳定性的问题。现在就是在基因组不稳定性这一块的研究上还有困难,不知道切入点在哪里,怎么来证明这一点。我们今天有免疫治疗,有突变的人对免疫治疗敏感,因为他抗原性比较强,形成了肿瘤特异性突变。实际上很多肿瘤,即使没有免疫治疗,也是有突变的活得好,活得长,不爱转移。我们要反思还有哪些内容我们没有关注到。
另外一个要说的是肿瘤的异质性。有人把肾癌不同的部位取材去测序;由于发生了转移,因此把其中的几个转移灶拿去测序。最后发现肿瘤组织中不同部位的突变谱不一样,说明这个肿瘤细胞有杂合性[8],很多突变存在于一部分病灶,不管是原发灶还是转移灶,有些突变存在于原发灶,有些存在于转移灶。只要这种变化不是在所有的肿瘤细胞中存在,它肯定不是在启动阶段发生的,即不是 driver,它可能是一种在肿瘤进展过程中因为基因组稳定性差形成的新变化,这种变化使得细胞获得了生长优势,而没有变化的细胞被淘汰了;如果相互之间没有明显的差别,生存优势差不多,它们就共存。
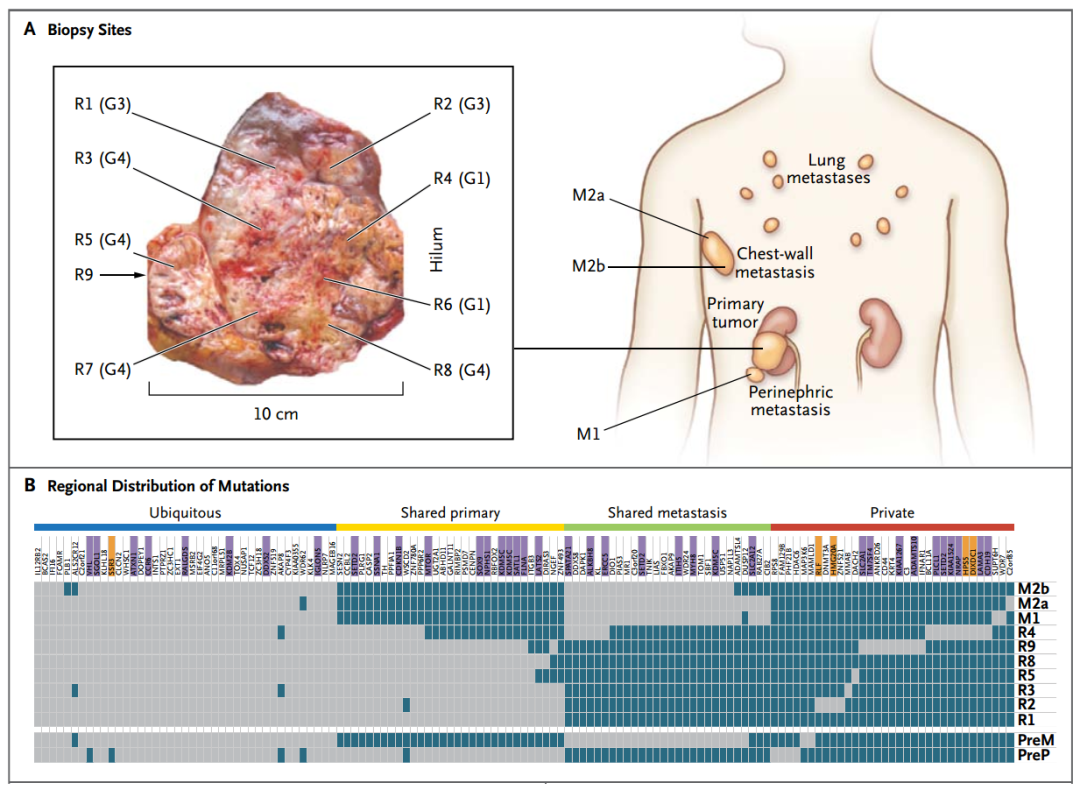
图 8. 多区域测序揭示肿瘤的瘤内异质性和多分支进化 [8]
这件事情告诉我们,要谈论一个细胞怎么发生癌变的,一定要看是不是在大多数细胞里面发生了变化。如果只关心肿瘤是怎么发生转移和耐药的,那么你关心部分亚群就可以了。很多人用 driver mutation、passage mutation 这些词,那是我们从肿瘤发生的角度来说,你要当心肿瘤的发生到底是不是突变造成的。
我们经常要考虑到的,到底是表观遗传更重要,还是遗传更重要,它们应该是相辅相成的。但很多情况下,表观遗传发生的比较早。其中有些表观遗传变化,会存在于每一个肿瘤细胞里,那你说它到底是不是驱动因素呢?还是只在某些肿瘤细胞里才是驱动因素?很多时候我们存在争论,从目前来看表观遗传可能有它很独到的地方,它在肿瘤研究上可以解释单纯用遗传因素解释不了的现象。我们在做胃癌的基因组学变化的时候,发现三分之一胃癌有 genetic 的变化,那剩下的三分之二是什么变化?我们要考虑有没有别的层次的指标,比如表观遗传。
OK,这里我们就讲完了肿瘤发生的多阶段性跟突变、driver mutation 的概念。
肿瘤的表观基因组学特征
胚胎化、去分化
我们刚才已经介绍了第一个特征,肿瘤的胚胎化、去分化。这个特征包括,肿瘤细胞去分化样的表观遗传变化以及宿主间质细胞发生了适应性的表观遗传变化。肿瘤间质细胞和正常间质细胞已经不一样了,它可以帮助肿瘤细胞生长。
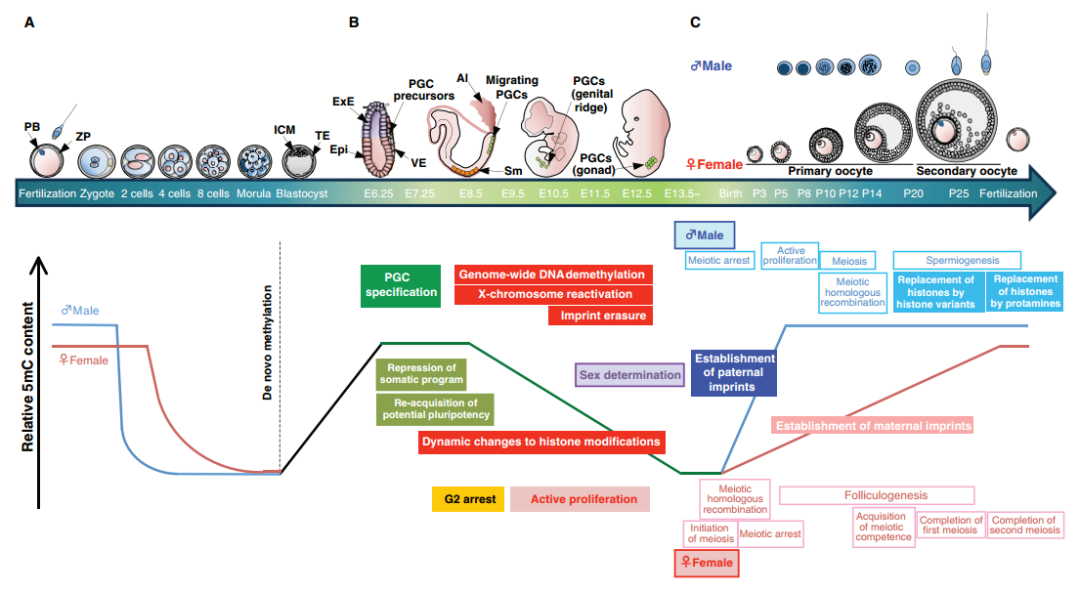
图 9. 哺乳动物生殖细胞和早期胚胎发育过程中 DNA 甲基化的动态变化 [9]
图 9 中可以看到,上面是细胞的生命周期,从一个受精卵到形成一个新的生殖细胞;下面是 DNA 甲基化的模式,精子、卵子形成合子,发育到桑椹胚时,除印记基因外大部分的 DNA 甲基化都被抹去。然后开始分化出不同的胚层,重现甲基化模式,再到机体的形成;在形成的过程中,新的胚胎发育,卵细胞、卵巢又重新形成,里面又有新的原始生殖细胞。
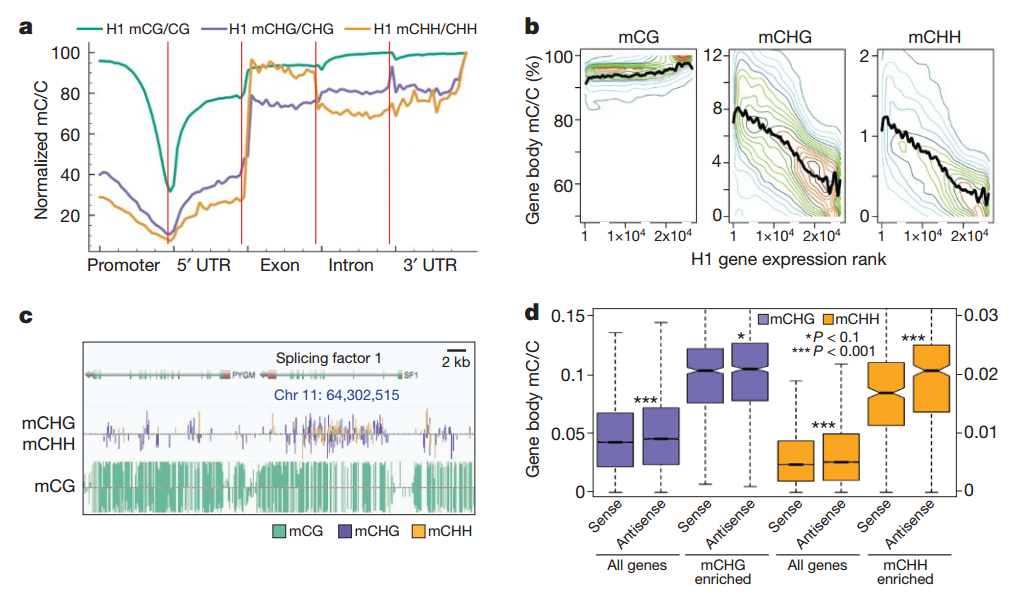
图 10. H1 人类胚胎干细胞中的 non-CpG 甲基化 [10]
肿瘤细胞也会呈现这些变化。举个例子,当我们谈论 DNA 甲基化时往往是说 CpG 位点,但在植物界、微生物和胚胎中,可以看到非 CpG 位点的甲基化,主要在 genebody 区(图 10)。在肿瘤组织中是不是也有这种变化呢?即非 CpG 位点甲基化与肿瘤细胞去分化的关系。到目前,我们还没有证据。为什么没有证据呢?这个要从我们 DNA 甲基化的检测说起。之所以 DNA 甲基化的研究会重新时髦起来,是因为出现了一个技术——重亚硫酸盐转化,它可以把没有甲基化的胞嘧啶进行化学转换,变成尿嘧啶。PCR 反应体系里面不加尿嘧啶,只加胸腺嘧啶,它就变成 C-T 转换了;而甲基化的胞嘧啶无法转换。但这个转换过程难以做到 100%,会有 1% 或 2% 的残留。当我们测出一个 non-CpG 位点的胞嘧啶还是胞嘧啶 C,没有变成胸腺嘧啶 T,你说它是没有转换成功,还是因为甲基化了呢?没有很好手段去鉴别它。但有人说,它很可能是非 CpG 位点的甲基化,只能说可能,不能说是板上钉钉的事。
肿瘤和组织特异性
第二个特征,肿瘤有肿瘤和组织特异性。
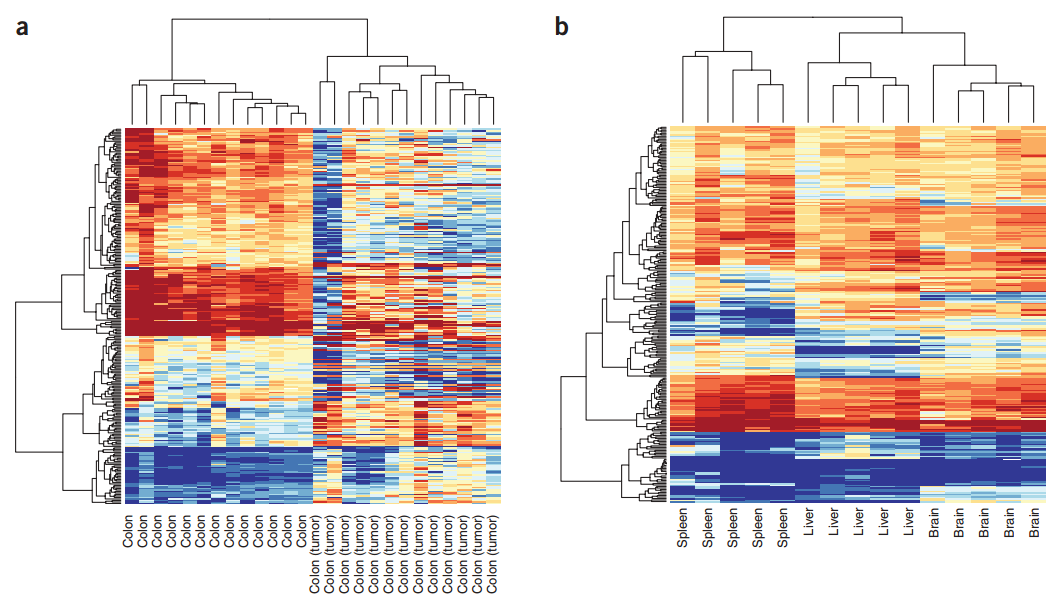
图 11. 正常组织样本及其肿瘤的特征聚类 [11]
图 11b 中有脾脏、肝脏、脑组织,可以看到不同组织间 DNA 甲基化的特征有一样的,也有不一样的,说明 DNA 的甲基化有组织特异性,准确的说也有细胞类型特异性。同一种细胞比如说纤维细胞,在不同的器官中,它的甲基化谱比较像,但也有不一样的地方。当肿瘤发生后,比如正常的结肠组织和结肠癌,你会发现它们彼此之间虽然有差别,但还是非常像(图 11a),这对于我们研究肿瘤具有很现实的意义。举个例子,大概 5、6 年前,我开始找胃癌转移相关基因,对于有淋巴结转移和没有出现淋巴结转移胃癌病人,我比较原发灶,看它们两者之间有什么差别。如果原发灶里面有很爱转移的肿瘤细胞,我就可以逮住它,以后就可用来预测肿瘤转移,我们后来把它做成功了[12]。但有一个日本的课题组,他比较的是原发灶和淋巴结转移灶,他觉得这两者之间差别应该更大,但他恰恰忘了,肿瘤细胞到淋巴结以后虽然也很像原发组织,但里面肯定掺杂了很多淋巴结的组织成分,产生了很多变化,而这些变化其实是淋巴结组织本身拥有的,或者是肿瘤细胞跑到淋巴结以后,为了适应淋巴结内环境,促使其发生的变化。他做了很多努力,但还是以失败告终。肿瘤组织跟它的正常组织之间很像,所以它们那些细微的差别才是肿瘤特有的。你要是比较不同组织间的表观遗传特征,只能说这两个组织之间有什么差别,不能用于肿瘤转移研究。
现在有很多人提出 “异病同治” 这么一个概念,尽管肿瘤长在全身不同的地方,但可能用同一种方案治疗。我不反对这个说法,但是 “异病同治” 这个方法首先就抹杀了肿瘤跟它起源组织之间的差别,我们对此要非常谨慎。
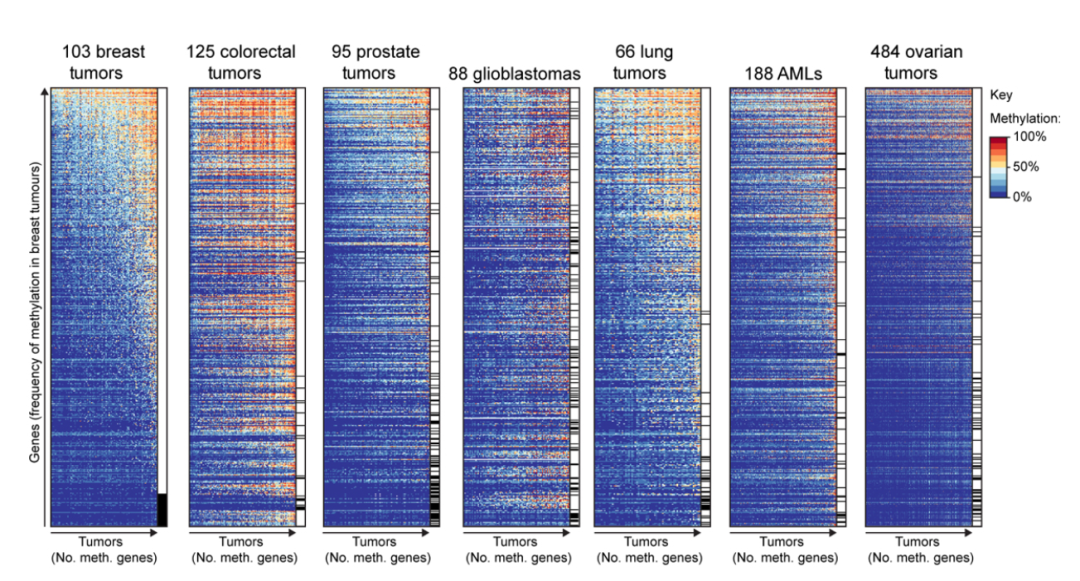
图 12. 肿瘤的组织起源决定了其组织特异性的 DNA 甲基化模式 [13]
另一个例子,来证明这件事情,肿瘤的组织起源决定了其 DNA 甲基化模式[13],正常组织变成肿瘤以后,它跟它原来的家庭出身是那么相像,永远抹不去。大家基本已经接受了这个理论。
上面我们有提到鳞状上皮的结构,上面是鳞状上皮,下面结缔组织;而在肿瘤组织里基本上见不到结缔组织了。结缔组织的表观遗传特征跟鳞状上皮的表观遗传特征有非常大的差别。当我们说肿瘤组织出现了某种正常组织没有的变化时,那可不一定是肿瘤特异性的,也可能像这样是因为结缔组织变少了造成的。这个时候要特别小心,我们要区分这个地方是组织结构造成的,还是肿瘤细胞里面发生的变化。当然如果你不关心间质的时候无所谓,如果你要关心间质,要么要间质和间质比较,上皮跟上皮比较。大部分肿瘤起源于上皮组织,上皮来源的肿瘤细胞和正常上皮两者之间有什么变化,这样比较才不会陷入组织细胞结构性变化造成的差异。肿瘤间质也是如此,正常间质是什么变化,肿瘤间质是什么变化,分别比较不会陷入这种问题。很多人说,我取一个肿瘤组织,再取一个切沿的正常组织,这个基因就有表达差异,但这个是肿瘤细胞造成的变化还是间质结构发生的变化,这个必须分清楚。这个是要借助于免疫组化或者是共聚焦鉴定,到底是肿瘤细胞还是间质细胞发生了这种特异性变化。不把这个事情做清楚,那些东西没有意义。
全基因组 DNA 低甲基化
然后再说全基因组 DNA 低甲基化。我们以前在做化学致癌的时候,用致癌物处理细胞或动物四到六个小时,全基因组甲基化水平一下子就降了很多,这是肿瘤非常显著的表观遗传特征;同时,基因组的稳定性就下降了。全基因组低甲基化和基因组稳定性显著下降,它们之间有没有内在的关系?这个问题到现在也没有搞清楚。如果它们是一回事,那很可能我们研究基因组不稳定性就找到了一个突破口。我们在做致癌的时候,根本不需要看几年以后的变化,就直接看几个小时以后的就 OK 了。怎么看?动物喝下致癌物后,你看它全身哪个组织发生了低甲基化,那个地方就是它的靶器官,未来很可能要长肿瘤,基本上就是这样。
低甲基化和全基因组不稳定性都是早期现象,那时候肿瘤还没有发生。这时候我们再回过头来,那可能是一种细胞的应急反应,发出 “SOS”。很久很久以前我们还是单细胞,每个细胞都是全能的,既要适应环境,还要繁衍后代。但自打我们变成多细胞生命体以后,细胞就分工了,除了生殖细胞还保留全能性,组织干细胞保留了部分的全能性,很多细胞的全能性被压制了。比如在一个群体中,组织运营很好,大家生活的很快乐;但是当环境迫使其中少数人不适合当前的生存环境,当然也可把它消灭掉;但万一没消灭掉,或者很多人都感受到不安全了,那就要造反了。“SOS 反应” 就是说,全基因组低甲基化也好,基因组稳定性降低也好,本质上都是说我在你这里活不下去了,我得独立自主,恢复我的全能性。我们每一个细胞,原来都拥有全能性,仅仅是为了不同组织的分工,牺牲了自身的全能性。当有致癌物,让你活不下去了,那肯定要造反,不服从分配了。我觉得肿瘤细胞,就是部分恢复了它的本来面目,部分恢复了它的全能性。所以全基因组低甲基化,是这里面值得研究的一个地方。
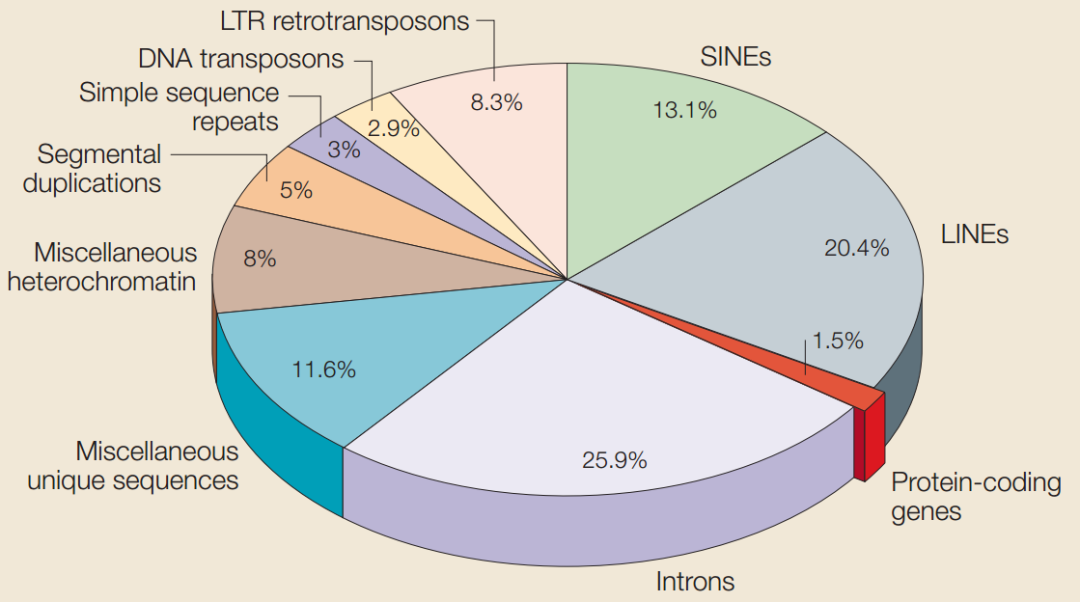
图 13. 人类基因组的成分 [14]
全基因组低甲基化发生在哪?以前没有基因特异性的分析方法,我们只知道甲基化水平下降了,但到底降在哪儿没有人去管。图 13 是我们的基因组的成分,1.5% 为蛋白编码基因,98.5% 的基因不编码蛋白;还有很多是重复序列,比如说 20.4% 是 LINE,LINE 是比较长的,散在分布的重复序列;还有 SINE,SINE 是比较短的,300bp 左右,每一个 Alu 中含有 24 个 CpG 位点,Alu 散在分布的重复序列。我们还有不是分散分布的重复序列,比如端粒,它是串联存在。LINE 和 SINE 占了我们基因组的三分之一,还有很多其他的转座子,我们都没去关注它。其中 SINE 里面有一种序列叫 Alu。Alu 序列是我们体内拷贝数最多的一个基因,我们每一个正常细胞中大部分基因是单拷贝的,但 Alu 有 160 万个拷贝。我们基因组里面三分之一的 CpG 位点都在 Alu 里。
所以说全基因组低甲基化到底发生在哪?是不是在这些重复序列里头,或重复序列起到了主要的贡献。很可惜,测甲基化的人很少关心它。早期用芯片时,一定要把 Alu 去掉,因为它是高度甲基化,会影响目的基因的观察。现在不管是 Illumina 450K 芯片还是最新的 850K,从来不关心它。现在我们回想一下,我们测出来有变化,病人就活得长;没变化的,活得不长。那是因为我们缺失了对全基因组不稳定性或全基因组低甲基化的关注,这件事情要重新引起关注。当我们发现蛋白编码的基因是如此之少时,我们要反思我们这么聪明,跟别的动物不一样,难道就靠这 1.5% 的编码蛋白?如果单看编码基因的多少,我们跟一只苍蝇差不多;但比较非编码区域时,它们肯定没我们多,比我们差远了。现在我们已经开始关注这些非编码区域,也陆续开始发文章。
Alu 序列是一种逆转座子,能通过逆转录反复地插入到基因组里面,功能上除了控制全基因低甲基化以外,还可作为核小体的定位装置。我们知道染色体是以核小体为基本单元的,核小体在基因组的位置基本上是固定的,尤其是转录起始点附近。没有转录起始点的地方通过 Alu 来定位,每一个 Alu 序列对应一个核小体,它散布在我们基因组里面,像停车场立的桩子,每个车位之间都要划线,但有的是三个桩子,有的是两个桩子,它往那个地方一占就停不了车。每一个 Alu 序列对应一个独立的核小体,它是核小体的定位装置[15]。
在 DNA 进行半保留复制时,双链解聚形成复制叉的地方是一个 Alu 序列。更重要的是, Alu 在有性繁殖过程中也发挥作用。不止我们人类,单细胞生物就开始了有性繁殖。为什么会这样?因为不同的两个生命体之间可以进行基因重组,可能获得了必不可少的生长优势。怎么重组?Alu 序列是一个重组位点,因为全基因组中有那么多序列相同的东西,跟你匹配马上就交换了。在显微镜下你去看,姐妹染色单体交换那个点肯定就是在 Alu 上。转座(translocation)的热点基本上也在 Alu 上[16]。我们年老后,基因转录水平降低,为了弥补蛋白质的量,谁来干这个事?Alu 会转录出 RNA 来,虽然它本身不编码蛋白,但可普遍增加蛋白翻译的效率。总之,我们对 Alu 作用的重视还不够。
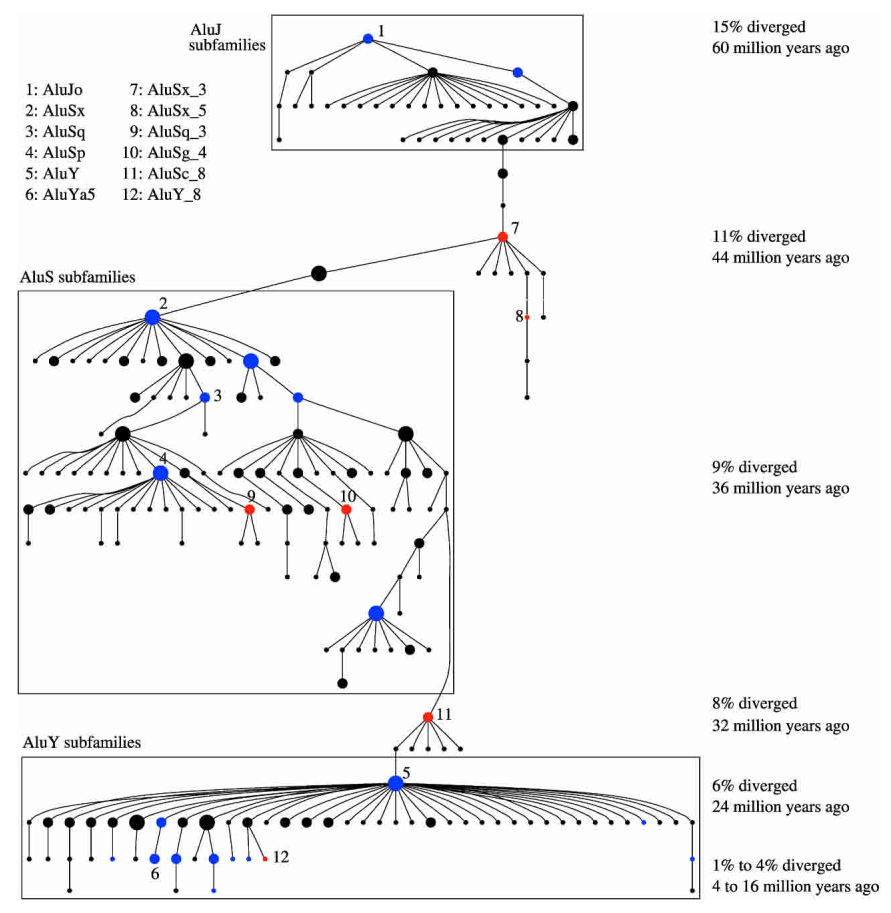
图 14. Alu 序列的演变过程 [17]
我们刚才提到基因中 1/3 的 CpG 位点在 Alu 序列中,但多数 Alu 发生了变化,某些 CpG 位点会发生脱氨基,在体内演变成 200 多种不同的 Alu(图 14)。尽管如此,在 Alu 里面 GC 含量仍然非常高,是非 CpG 岛区的 8 倍,这说明一件什么事情呢?我们基因组中的胞嘧啶,如果无关紧要,它很可能会脱氨基就变成胸腺嘧啶,这也是为什么大部分突变发生在胞嘧啶上,这会进化出新的基因来,是基因进化的动力。为什么 Alu 序列里的 CpG 位点胞嘧啶没有脱氨基,而是进化过程中被保留下来?没有脱氨基的 Alu 才能传递下去,这说明了甲基化的重要,也提示 Alu 不是简单的重复序列,它是有功能的。
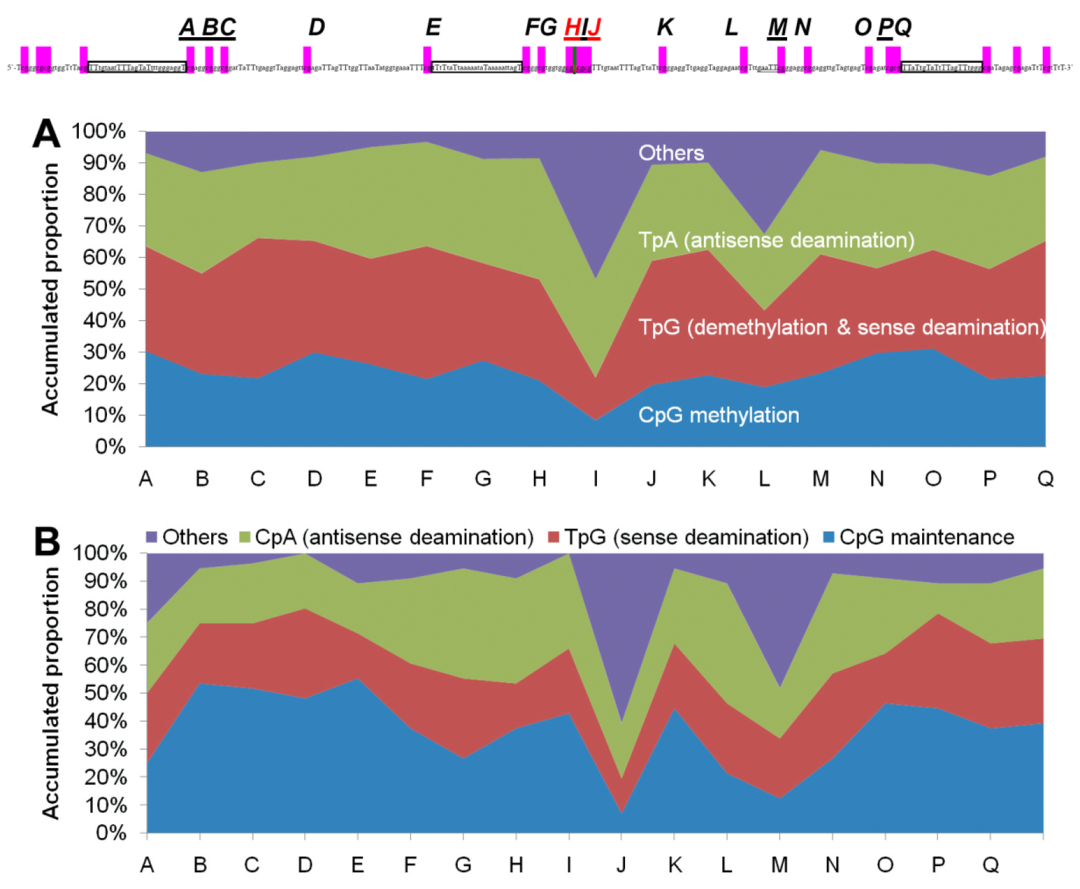
图 15. Alu 元件的胞嘧啶脱氨基化 [18]
早期很多人测 Alu 的甲基化,总是看不到很好的结果,主要原因是分析 Alu 序列的方法不成熟,大家忽略了 Alu 真实的甲基化位点在哪里。图 15 最上方是一个标准的 Alu 序列里面的 24 个 CpG 位点,有些已经突变了脱氨基了,但是总的来看比如 I 位点基本上全突变了,没剩下多少维持下来,甲基化很低;而 A、D、O 这三个位点,还保存在那里,大部分甲基化。用这个方法来测体内 Alu 甲基化情况可能会更准确,整体仍看不到什么好的结果,我们觉得考虑应该具体到基因,即基因组中哪一段 Alu 序列变化会影响生命的过程,影响基因的表达模式,这样研究才会有意义。这已经超出了我们研究单个基因的表观遗传修饰的概念,这是从基因所处环境控制基因表达的角度研究。当我们把 1.5% 的蛋白编码基因理解成种子,把其余 98.5% 的序列理解成土壤,从这个角度才能解释这些非蛋白编码区的低甲基化状态改变如何影响细胞的生命,如何使得细胞生存下来。不管是同源重组还是非同源重组,突变是进化的动力;而肿瘤为了生存下去,要演变出很多突变,这从逻辑上解释得通,但需要提供证据——哪些位置低甲基化了,突变率就变高,而逆转这个过程是否就不发生突变了。
CpG 岛甲基化状态变异
我们研究比较多的是转录起始点附近的 CpG 岛,主要位于启动子区。我们常说的启动子甲基化往往指 CpG 岛甲基化,但不排除第一外显子,有的 CpG 岛就是跨第一外显子的。我们对非启动子区甲基化研究很少,在第二外显子第三外显子等相对保守的区域,也有 CpG 岛存在,但它们有没有功能?是否影响染色体的构象或者是否会影响基因剪接、染色质重塑过程?这些还不清楚。
一个基因要表达,转录起始点附近的 CpG 岛(一两个核小体范围内)不能甲基化;如果甲基化,就转录不了,没有例外。有些人说我拿一个组织,在特定位点测出了甲基化,但发现 RNA 转录了,这是犯了概念性错误,当我们说一个启动子甲基化了,基因不能转录,那是指在同一个分子上;而描述一块组织时,一个组织里有成千上万个分子,有些甲基化了,而另一些没有甲基化但可以转录,即使检测到转录出的 RNA 也不能说明什么。一般来说,研究 DNA 甲基化是否影响基因的转录活性用细胞系比较好,虽然不是完全均一,但相对来说比较均一,代表性会比较好。一些单细胞组学等新技术出来后,可能会修改一些看法。很多那时候在组织中也可以看到相关性,但没有看到相关性也不能否定甲基化对转录的抑制作用。其意义在于在 DNA 水平上我们研究基因的可转录性,而不是真实的转录情况,因为一个基因要转录除了没有甲基化外还需要转录因子辅助才可以,未甲基化提供了一个可转录的状态。
我们都知道 DNA 非常稳定,在血和毛发甚至化石中都可提取 DNA。因为 DNA 甲基化通过 α- 键共价结合,跟 DNA 一样非常稳定,因此 DNA 甲基化的检测对样本的要求很低。此外甲基化序列与非甲基化序列可以用不同的方法分别分析,互不干扰,因此可以对少数细胞的变化进行准确灵敏的分析。
羟(去)甲基化能力降低
肿瘤里有一个特征,羟甲基化大幅度下降。而现在的 DNA 去甲基化通过糖苷酶切除一段 DNA,又重新复制和连接回去,是一种极其浪费的模式;最简单的是把羧基脱下来,直接一步完成。受精卵分裂成八细胞的过程中,把所有的甲基化需要全抹掉,羟甲基化也在其中,如果这样切,双链 DNA 会切成碎片,事实上不是这样的,如果这种核苷酸切除修复途径真存在,也是单链上进行,不会两条链同时进行,但还没证据。我在 2014 年的综述《DNA 甲基化和去甲基化的研究现状及思考》中专门讨论了这个问题[19]。
在肿瘤组织中,之所以会羟甲基化水平降低,可能是 TET1 自身发生甲基化而失活,也有可能是 TET2 发生突变。我们自己的工作发现,用常规的分析方法,P16 甲基化和羟甲基化在早期与晚期肿瘤之间没有差别,但是把羟甲基化与单纯甲基化分开,每个都有差异,提示有些羟甲基化标志在肿瘤中可以保留下来。
肿瘤表观遗传标志物
表观遗传修饰有很多,但直到今天能往肿瘤标志物上靠的只有 DNA 甲基化,因为研究的技术手段成熟,还有上面提到的一堆优点。组蛋白修饰确实很重要,但目前的研究方法主要基于抗体,把某种修饰的组蛋白拽下来,然后提取 DNA 进行 PCR 扩增,判断某个位置的修饰情况。用抗体的结合是一种间接的证据,并没有直接看到某段核小体上组蛋白是否发生修饰。目前组蛋白修饰还难以作为肿瘤标志物,顶多推测一下某种组蛋白总体的变化跟肿瘤转变的关系。大约 20 年前,miRNA 成为明星分子,大家对它期待很多,期待最多的是 miRNA 能否导致表型变化。是不是吃下去 miRNA 就能把某个基因关掉,就能把病治好,但至今没有听说过类似研究,只是有些研究表明 miRNA 局部渗透到达一些细胞里可以干扰基因的表达变化。虽然有科学家说,植物的 miRNA 可调控人体细胞基因的表达,甚至说那可能是一种营养素,我不认同这一点。
今天讲表观遗传标志物时只讲 DNA 甲基化。表观遗传标志物可以用来干什么呢?我们刚才介绍了肿瘤发生的多阶段性,不同阶段有不同的用处。
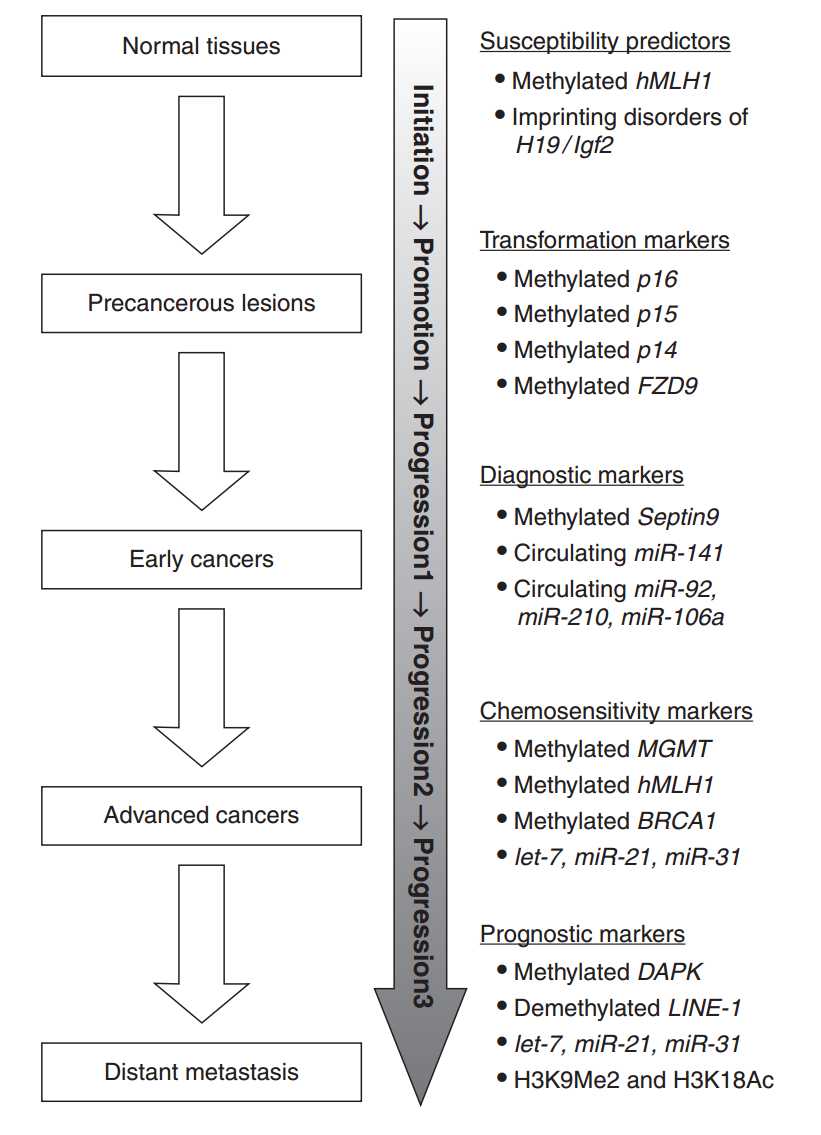
图 16. 肿瘤发展多阶段过程中不同的表观遗传标志物 [20]
从正常组织转变为癌前病变的过程中,标志物可以看出你对致癌物是否易感,可以指导我们应该以哪种方式生活,可不可以抽烟喝酒,可不可以去有毒环境下工作,可不可以去高原生活等。如果已经有癌前病变,标志物可以判断哪些可以变成癌哪些不会发展成癌,可以通过检测血液尿液或粪便来发现早期肿瘤。我们有很多方式治疗早期肿瘤,早期肿瘤的生存率比较高。到了晚期肿瘤,这些标志物可以提示我们用什么药物治疗,哪个药物有效。所以,无论哪个阶段,表观遗传标志物都有用武之地。
癌前病变癌变预警标志物
第一阶段的标志物是癌前病变预警标志物。
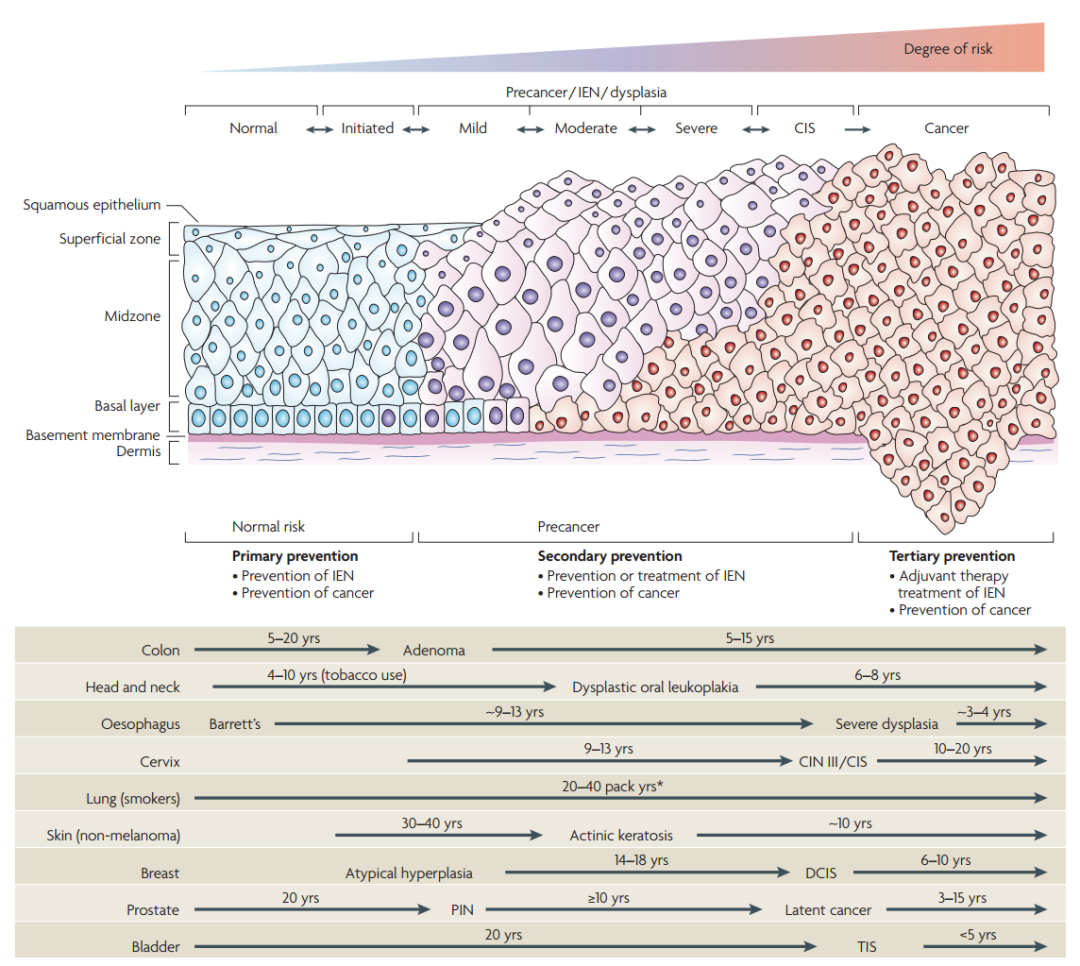
图 17. 肿瘤发生的多阶段 [21]
什么是癌前病变呢?在正常细胞变成癌的过程中有一个异型增生阶段,即癌前病变。80% 恶性肿瘤起源于上皮,它们的共同特征就是癌前病阶段,显微镜下可以看出来的,癌前病变一出现意味着患癌风险比正常人高了 100 倍,但是显微镜下看不出来谁会癌变,谁不会癌变,只有 1/3 会癌变,2/3 不会癌变甚至会消退。显微镜下不能断定谁会进展,谁会稳定,谁会消退,只能过度治疗。比如宫颈异常增生,发现后通常会切了,反正生了一个孩子就再也不生了,但今天政策变了可以生二胎了,那还可以切吗?在日本胃癌很多,他们不害怕做胃镜,几年做一次,胃镜如果看到异型增生,就会选择切掉,这会造成一种人工溃疡,要忍受两年疼痛才会基本愈合,这也是过度治疗。因为没有办法判断癌前病变是否进展,所以把它当作癌来对待。如果癌前病变有一部分恶变了,那么其 DNA 甲基化肯定跟正常细胞不一样,把这些变化能准确测出来,意味着可能可以预测哪些会癌变。
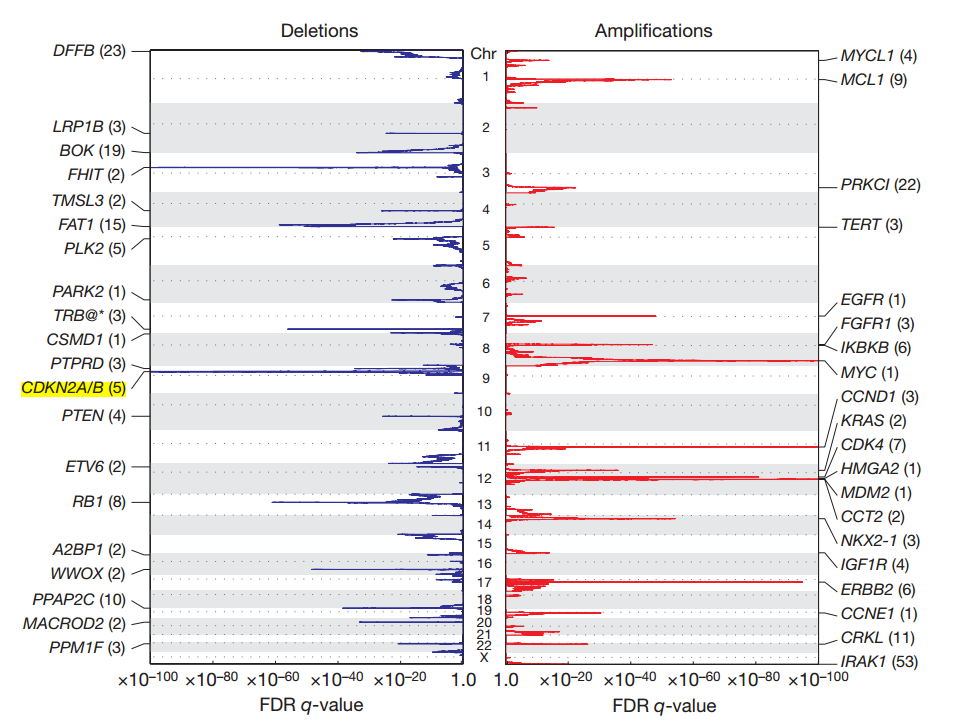
图 18. 3131 份肿瘤组织中基因拷贝数的变异 [22]
2010 年的一项研究对人体 3131 个肿瘤切片进行拷贝数测定 [22],图 18 中左边是 deletion,基本上都是肿瘤抑制基因;右边是 amplification,基本上全是癌基因。我们看一下 P16 (CDKN2A/B) 基因所在的位点。P16 基因座上不只有 P16,一个基因编码 P14 和 P16 两个蛋白,可以看到它在肿瘤组织中缺失最多。P14 和 P16 共用第二第三外显子,P14 和 P16 两条通路控制了 RB 的磷酸化,并控制了细胞 G1/S 期转换。在 G1/S 期转换时,模板必须完美无缺才能开始复制,如果有缺陷就不能进入半保留复制,要么修复要么凋亡;如果不凋亡,而且还进行了半保留复制,就变成了一个缺陷的细胞。如果异形增生组织中,如果非正常的细胞可以无限增殖,必然发展为肿瘤。P16 基因缺失频率是最高的,大概 10%,甲基化是否是它最主要的失活途径?所有肿瘤中有 30% 的病人中 P16 甲基化异常,是基因缺失频率的三倍,我们该怎么利用预测癌前病变呢?
在胃癌高发区中,胃癌的发生率是 10 万分之及六十几,而在低发区是 10 万分之十几,我们提出的问题是胃癌发生会不会存在癌前病变。当时我们在山东招募了 4000 人,都是 35 岁以上,五年进行一次胃镜,最后其中 100 多人得了胃癌,而胃癌基本上都是从癌前病变演变过来的,所以那一阶段的工作确定了癌前病变确实会发展成癌或者癌起源于癌前病变。通过这个工作,才敲定了这个理论,并且给我们提供了无价之宝。我们既有癌组织,又有几年前的癌前病变组织,如果说发现了某个在临床上使用的标志物,那么应该可以在临床前瞻队列中得到验证。
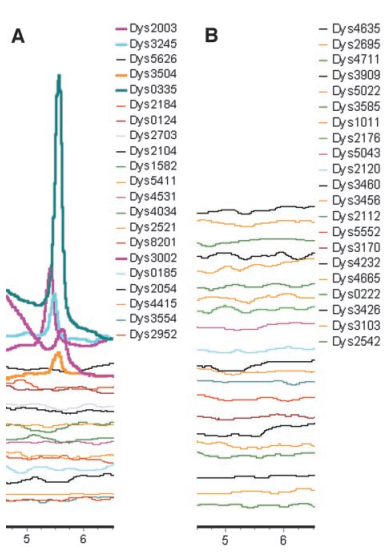
图 19. 胃粘膜异型增生样本中的 P16 甲基化 [23]
在 2004 年的研究中我们发现一组病人可以根据癌变部位可以找到原发灶,而另外一组病人癌前病变部位一样,但没得癌,在显微镜下看不出差别。但检测 P16 甲基化,癌变的一组可以看到甲基化的存在(图 19A),没癌变的没有 P16 的甲基化(图 19B),有甲基化的 5 个人(图 19A 中 P16 甲基化出现峰值的五个病人)都成了癌,这提示 P16 甲基化可能作为一个癌前病变的标志物。
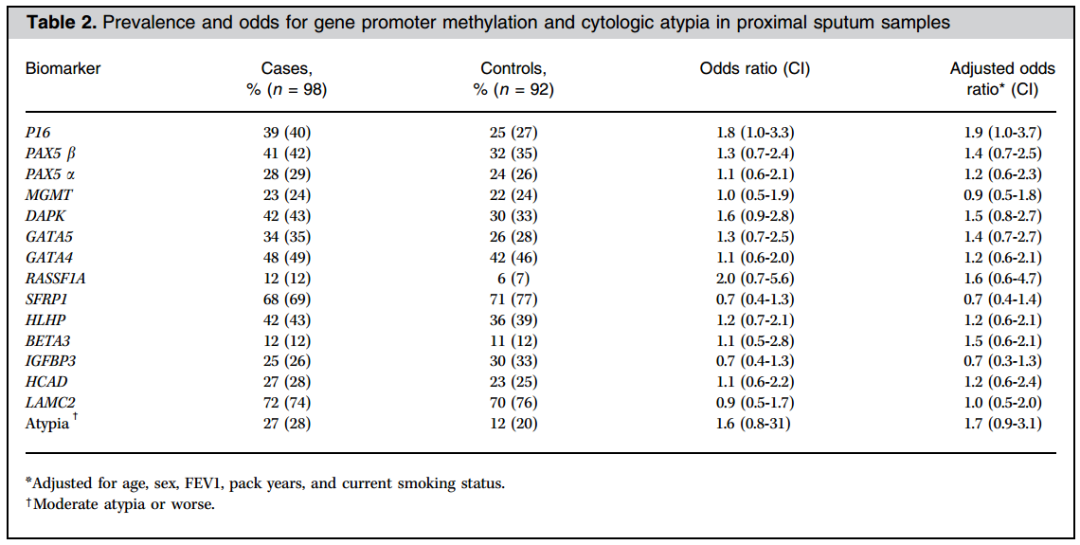
表 1. 痰样本 P16 甲基化与肺癌发生 [24]
2006 年约翰霍普金斯大学的研究者进行了另一个巢式病例前瞻性队列研究,把吸烟男性的痰收集起来,看谁得癌了,然后把三五年前的痰拿出来测一组基因的甲基化,发现了痰样品中 P16 甲基化与肺癌发生的关系,测了一组基因,P16 基因甲基化的差别很显著[24]。这个大学的另一组人,发现 P16 甲基化与 Barrett’s 食管癌变的有关[25]。这些研究说明 P16 甲基化确实可以用来做癌前病变标志物。
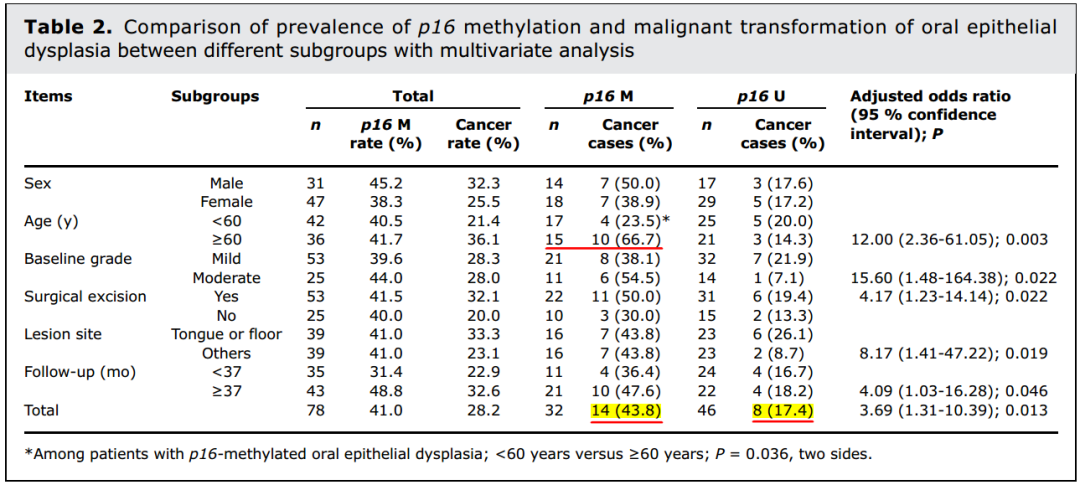
表 2. P16 甲基化与口腔粘膜上皮异形增生癌变前瞻队列研究 [26]
我们做了口腔粘膜上皮异型增生癌前病变的前瞻性研究,主要是因为口腔粘膜易于随访观察。我们招募了 200 个癌前病变病人,根据 P16 甲基化分成两个队列,随访五年看两组癌的发生率,可以看到有 P16 甲基化组癌症转化率(43.8%)大约是无甲基化组(17.4%)的 2.5 倍(表 2);大于 60 岁的老人里,15 个有甲基化的人中 10 个癌变,占 2/3;这提示 P16 甲基化确实可能是一个标志物。我们接着在临床检测方法上下功夫,首先要知道 P16 主要分布在哪些地方,是否稳定,还要建立分析方法。
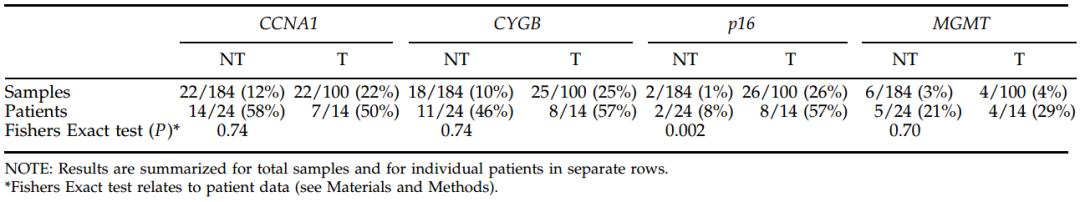
表 3. 异形增生未转化(NT)和转化(T)样本中基因启动子的突变频率 [27]
在后续的研究中,我们发现 P16 甲基化的效率相当于 AFP 筛查肝癌的效率,我们有点不敢相信。一个英国的研究组做了四个基因的甲基化,发现只有 P16 有变化,灵敏度和特异性都比较好,验证了我们的实验结果[27]。我们对我们的研究和他们的进行了比较,发现敏感性和特异性相近,有两个独立的研究者得到类似的结论,我们就比较放心了。
这个时候我们看 P16 基因在体内到底是怎么甲基化的以及甲基化状态是怎么分布的。在 400 多例胃癌组织中,把有 P16 甲基化的全部进行克隆测序,看甲基化分布位点。在炎症组织中也可以看到 P16 甲基化,这解释了为何幽门螺杆菌感染后出现很高的 P16 甲基化,但这种 DNA 不在 CpG 岛。而在肿瘤组织中,其 P16 甲基化就过渡到了启动子区域。而且在 5’-UTR 和第一外显子的核小体区域的 DNA 甲基化不稳定,TSS 附近的 DNA 甲基化非常稳定。

图 20. 人类胃癌组织中的 P16 CpG 岛甲基化情况 [28]
此外,我们还建立了 DNA 甲基化的检测方法[29],并且生产了试剂盒,随后进行了多中心的双盲的前瞻队列研究[30],也得到了类似的结果。在患者在口腔中看到白斑以后,大夫推荐做活检,看是癌前病变还是非异形增生癌前病变。如果一个病人看到了口腔粘膜白斑异形增生,由于可以看到,病人可能会每天看,自己吓自己,可能都没有办法正常工作了。但只有 1/3 的人会发生癌变,2/3 的人没事,如果你能告诉病人你最后会没事,那么你是积了多大的功德。
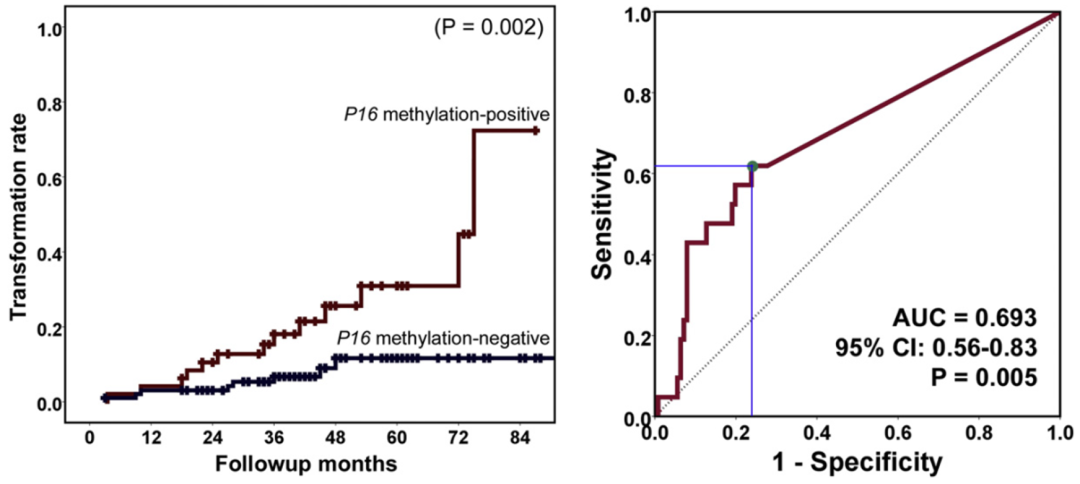
图 21. P16 甲基化阳性和阴性的口腔粘膜异形增生中的肿瘤转化率和肿瘤进展预测 [30]
在多中心的队列研究中,发现没有 P16 甲基化的病人中,只有 18% 的癌变率;而有 P16 甲基化的人,最终 78% 的病人出现癌变[30]。这些结果最终确认 P16 甲基化跟口腔粘膜异形增生癌变有确定的正相关的关系。
肿瘤早期筛查标志物
肿瘤早期筛查标志物有一个比较成功的例子,这个标志物也走了很漫长的路,大约 2000 年我们刚开始做表观遗传的时候德国研究者成立了一个 epigenomics 公司,就想做甲基化试剂,就做 SEPT9 这个基因,最开始是做这个基因的表达调控与肿瘤的关系,但最后他们以公司的模式做,不是以科学家的模式做,就朝这开发标志物开发产品去了。他们最后发现这个基因第五个外显子附近外显子有一个 CpG 岛,这个 CpG 岛的甲基化跟结肠癌有关,而且可以从血液中检测出来,所以他就卯足了劲要把这个开发成试剂盒。
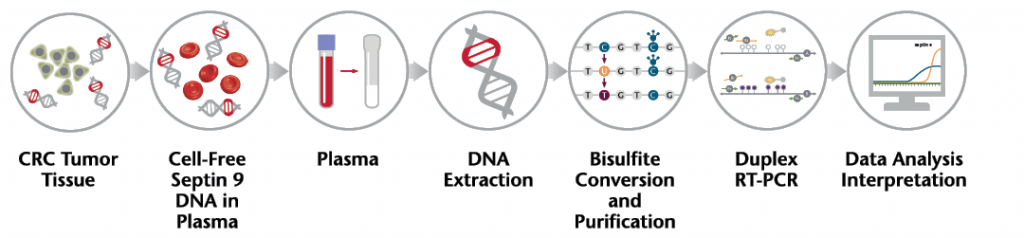
图 22. Epigenomics 公司 Epi proColon® 2.0 CE 筛查结直肠癌的工作流程
图源 https://www.epiprocolon.com/en/laboratories/
2010 年他们就鉴定了 SEPT9 这个方法,但他的试剂盒五年以后获得欧洲授权,2016 年获得美国授权。这个诊断试剂盒现在全球都在用,为什么呢?很多人不愿意去做结肠镜,但在血里面测出来甲基化的标志,难道你还不去做它吗?早期筛查物中多少年来很少出现一个好用的,而这是一个。
肿瘤预后标志物
预后标志物,是指示肿瘤是否会复发、转移、患者会不会死的很快或根本死不了的一类标志物。如果肿瘤会发生转移,或手术后发现发生了转移,与没有转移相比,我觉得一定有肿瘤转移干细胞的存在,而且数量会有差别。于是为了研究这件事,我们就测转移的胃癌和不转移的胃癌的甲基化组,比较原发灶和转移灶。组学筛查非常容易,做完这个结果后我们都傻了,发现了有非常多的差异。不只我一个人傻了,很多人这个时候就会找几个基因做一做,看看有没有什么功能别人以前没有报道过,发篇文章就结束了。但那样的话永远都发现不了标志物,我们傻傻的做了这样一件事:对于差异最大的,通过文献分析是否和肿瘤有关,有关就拿出来一个一个验证。三个博士(两个三年制,一个五年制的),在癌组织、癌旁切沿组织和 50 个非肿瘤病人的黏膜组织中,每个人做了 100 个基因,包括 78 个蛋白质编码基因和 10 个 miRNA 编码基因,然后就去比较,发现有 15 个基因(BMP3, BNIP3, CDKN2A, ECEL1, ELK1, GFRA1,HOXD10, KCNH1, PSMD10, PTPRT, SIGIRR, SRF, TBX5, TFPI2, ZNF382)的变化和肿瘤发生相关。有些可以进一步地筛查,其中有 3 个基因(GFRA1, SRF, ZNF382)和肿瘤转移相关。做完以后我们就做了一个发现队列和一个验证队列,还找了一个韩国和一个日本的验证队列,一共做四个验证队列[12],现在已经在做前瞻队列研究了。
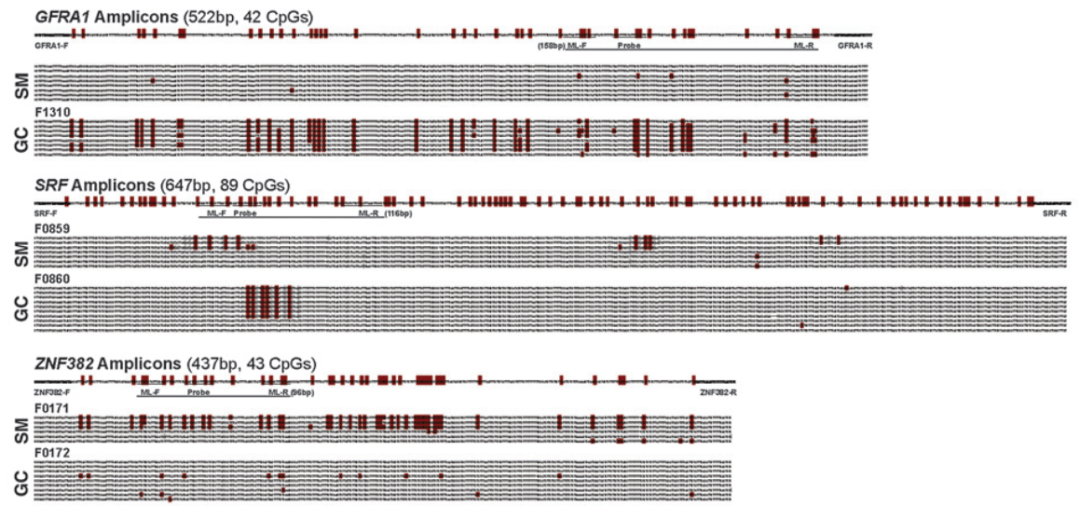
图 23. 与胃癌转移相关的三个基因的甲基化情况(GC,胃癌;SM,切沿)[12]
这三个与转移相关的基因,在不同的队列里都与转移侵袭有密切的相关性,然后鉴定甲基化位点。GFRA1 基因在正常组织中是甲基化的,在肿瘤组织中也表达,不过去甲基化了。这三个基因在发现队列和验证队列中非常一致,对各个队列中选取 200 个人,做了三年随访,发现其中两个基因确实能够预测癌症。但是它能否成为一个肿瘤预测的标志物,不只取决于我一个人,还取决于社会的接纳程度。
疗效预测标志物
举一个很典型的用在胶质瘤疗效上例子。鸟嘌呤第 6 位甲基化以后,就会突变,基因的突变很难修复;只有 MGMT 酶能把它修复了。这个酶可以修复两个鸟嘌呤的甲基化, MGMT 结合到甲基化的鸟嘌呤并修复以后,它自己就降解了。对肿瘤病人来说,这个酶最好是不工作的,然后烷化剂(比如替莫唑胺)穿透血脑屏障,可治疗神经胶质瘤和转移性的脑肿瘤,有点效果,但前提是这个人的鸟嘌呤第 6 位必须是甲基化的,如果是没有甲基化的,就没效[31]。这提示鸟嘌呤的甲基化状态可用于预测治疗的疗效。
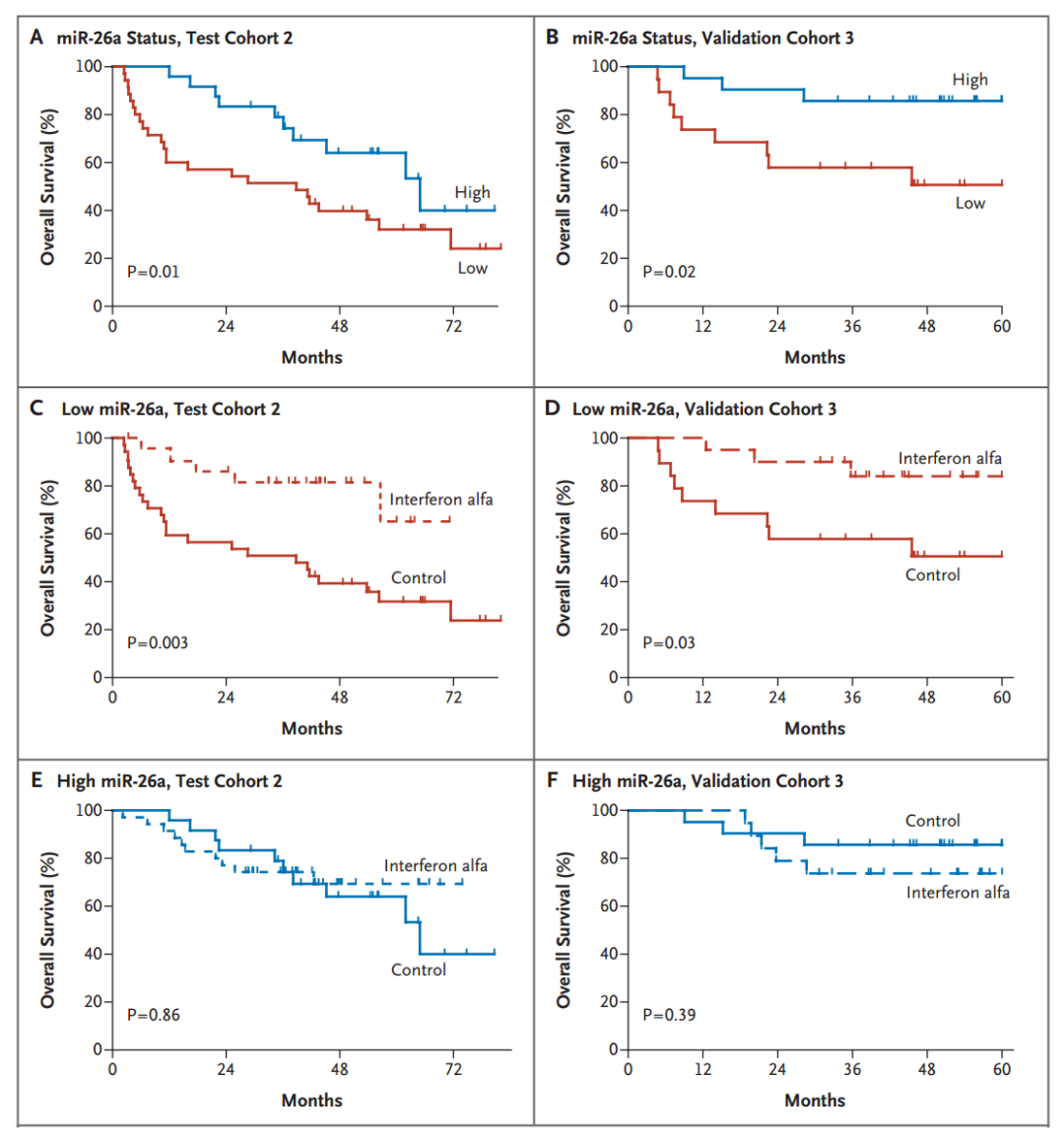
图 24. miR-26a 可用于区分α- 干扰素对肝癌的疗效 [32]
另一个表观遗传标志物——miR-26a 也可以用来预测疗效。乍一看发现 α- 干扰素治疗肝癌病人会有效,发现队列和验证队列都有效。仔细一看,你就会发现,只有对 miR-26a 表达很低的病人用 α- 干扰素治疗才有效,不管是发现队列还是验证队列;当 miR-26a 表达量很高时,没有效果[32]。这类标志物很大的价值就在于在临床上,对于大量的肿瘤患者,可以判断什么时候用什么药。
肿瘤的表观遗传治疗
最后一部分我们讲解表观遗传的治疗。现在已经出现了很多表观遗传药物,很可惜早期的 DNA 甲基化阻断剂比如 5-Aza-C 已经被发现了几十年,它除了干扰 DNA 甲基化,还能干扰了 mRNA 的合成,所以毒性比较强。后来就进一步发展为 5-Aza-CdR(商品名,地西他滨 decitabine),不会干扰核酸,只干扰脱氧核糖核酸。最近发现低剂量的使用才是 5-Aza-CdR 发挥作用最好的方式,已批准用于治疗骨髓增生异常综合征(Myelodysplastic syndromes,MDS),而且跟化疗药物组合在一起可能有比较好的疗效。
刚才我们提到组蛋白修饰没办法用来做肿瘤标志物,但组蛋白去乙酰化的药物一批一批的出现。其中西达苯胺(Chidamide)是中国研发的药物[33],已经在中国和美国上市,主要应用在外周 T 淋巴瘤上。还有一种是针对 EZH2 的药物,EZH2 是催化 H3K27me3 的一个蛋白,它也有抑制剂,但能否走向临床还不知道。SIRT1 的激活剂白藜芦醇[34],是葡萄酒中的抗氧化剂,也是一个表观遗传调节剂,但能不能作为一个治病的,值得深入的研究。
2015 年有一个令人兴奋的研究。现在肿瘤的免疫治疗很火,这项研究发现 DNA 甲基化阻断剂还有一个奇特的作用。我们的基因组里有很多逆转录病毒整合进去,外源整合的 DNA 通过甲基化沉默掉,但 DNA 甲基化阻断剂一上去就发现,这个逆转录病毒整合的基因去甲基化了,被激活了,能够表达出 RNA 来;而细胞会识别它,就会对干扰素变的易感[35],所以这是 DNA 甲基化阻断剂给肿瘤免疫治疗创造机会的一种机制。
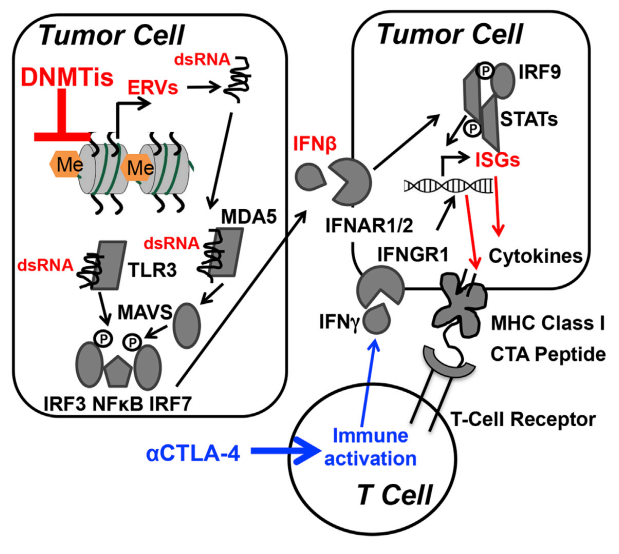
图 25. DNA 甲基化抑制剂通过激活逆转录病毒增强肿瘤对免疫治疗的敏感性 [35]
逆转录病毒元件不仅存在于肿瘤细胞中,也存在于我们体内的每一个细胞中,那么这个甲基化阻断剂进去后把每一个细胞的逆转录病毒整合基因都激活了,那么是不是敌我不分,杀死肿瘤的药物也会把正常的组织杀死?当然我们可以改良,可以局部注射,减少对全局的损伤,但这种途径至少创造了一个机会。
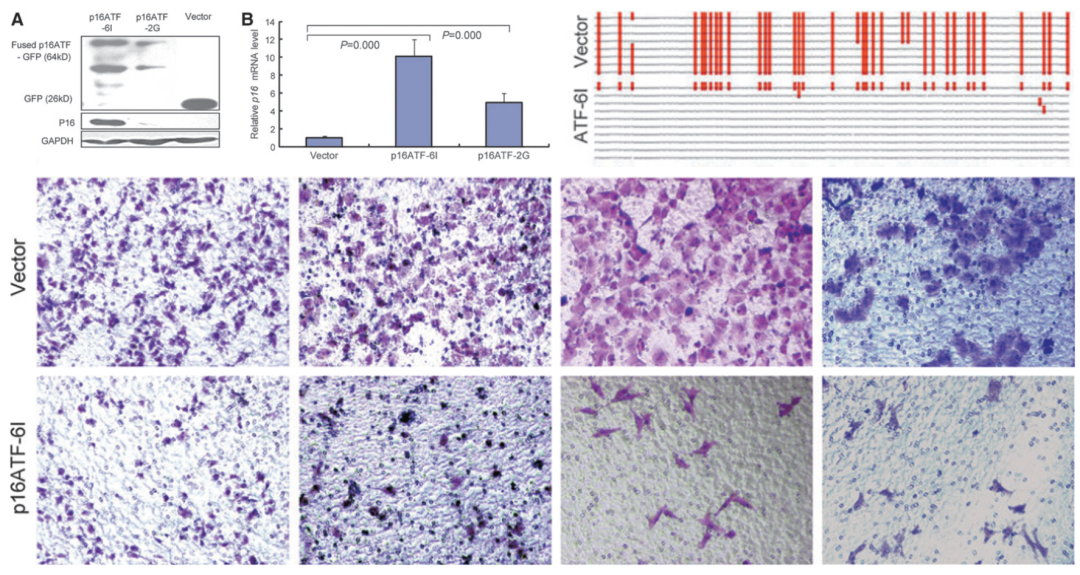
图 26. 特异性的 P16 基因去甲基化激活 P16 表达,抑制肿瘤侵袭和转移 [36]
那么这是我们自己的一个例子。我们都知道 DNA 甲基化不是一种突变,可以被抹掉,除了 DNA 甲基化阻断剂阻断以外,我们可以采用基因特异性的去甲基化的方法。用 5-Aza-C 的时候,没有特异性,把全基因组甲基化全抹掉了,肯定是有毒性的。但如果我认为这个甲基化非常重要,做一个基因特异化的 DNA 甲基化,就奔着这几个基因去了。首先设计了一个锌指蛋白,可以特异性结合到 P16 基因上去,然后把它和 VP64 结合到一起,加上核定位信号;构建的载体进入宿主后入核,确确实实发现这些基因被激活了,共聚焦可以看的很清楚,激活以后抑制了细胞的侵袭和转移[36]。利用这个我可以做一个什么事呢?口腔黏膜一般我可以贴膜,口腔溃疡有贴膜的治疗方式。是不是我这个东西贴上去也能把它激活呢?就用这么一个简单的方法,做了这么一件事。
还存在一个鸡和蛋的问题。有人说,DNA 甲基化是伴随着基因沉默发生的现象,那么它自身在基因转录上到底有没有功能呢?所以我们就是觉得我们手上有这么一个基因特异性结合蛋白,我们就把这个 P16 基因构成了一个特异性甲基化酶(DNMT3a 的催化结构域),转染细胞发现 P16 基因表达降下来。P16 甲基化以后,细胞的迁移能力变快了(增殖不受影响)。将 P16 甲基化的细胞注射到小鼠尾静脉,发现无论肿瘤变大,肿瘤转移会加速。我们这个结果回答两个问题,第一,P16 甲基化是直接抑制基因的转录;第二,P16 的甲基化促进肿瘤侵袭和转移[37]。
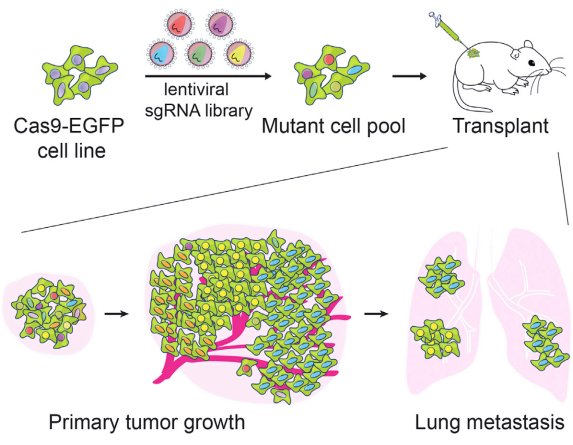
图 27. 在小鼠肿瘤生长和转移模型中全基因组 CRISPR 筛选策略 [38]
最后分享一项很受启发的工作,来自张峰课题组。他们设计 67450 个 gRNA,然后包装成逆转录病毒库去感染细胞,理论上每个细胞内会有转入几种 gRNA,相应地几个基因会被失活。然后把这些细胞混到一起,然后种到小鼠身上,看长出来的肿瘤里面到底哪个基因失活,最后发现一个是 Pten,一个是 p16,就是在转移性癌细胞里面大多带有这两个基因的突变,提示这两个基因有促进肿瘤生长和和转移的作用[38]。以前没有人敢说 P16 基因可以促进肿瘤的转移,但事实就放在那。而我们的工作告诉大家,P16 的甲基化也有类似的作用。这就是我们在肿瘤治疗上做的一点工作,又做了一点功能。就是这些,谢谢大家。
文稿整理 | 澳门科技大学 彭宇中,中山大学肿瘤防治中心 陈梦珂,中科院遗传与发育所 战珍萍
全文统筹 | 复旦大学生物医学研究院 徐鹏
参考文献:
- Bell, R.B., P.E. Andersen, and R.P. Fernandes, Oral, head and neck oncology and reconstructive surgery. 2017.
Hanahan, D. and R.A. Weinberg, Hallmarks of cancer: the next generation.Cell, 2011. 144(5): p. 646-74.
Kim, J.H., et al., Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson’s disease. Nature, 2002. 418(6893): p. 50-6.
Correia, A.S., et al., Stem cell-based therapy for Parkinson’sdisease. Ann Med, 2005. 37(7): p.487-98.
Knudson, A.G., Two genetic hits (more or less) to cancer. Nat Rev Cancer, 2001. 1(2): p. 157-62.
- Weinberg, R.A. and R.A. Weinberg, The biology of cancer. 2013: Garlandscience.
Kandoth, C., et al., Mutational landscape and significance across12 major cancer types. Nature, 2013. 502(7471):p. 333-339.
Gerlinger, M., et al., Intratumor heterogeneity and branched evolution revealed by multi region sequencing. N Engl J Med, 2012. 366(10): p. 883-892.
Saitou, M., S. Kagiwada, and K. Kurimoto, Epigenetic reprogramming in mouse pre-implantation development and primordial germ cells. Development, 2012. 139(1): p. 15-31.
Lister, R., et al., Human DNA methylomes at base resolution show widespread epigenomic differences. Nature, 2009. 462(7271):p. 315-22.
Irizarry, R.A., et al., The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet, 2009. 41(2):p. 178-186.
Liu, Z., et al., Large-scale characterization of DNA methylation changes in human gastric carcinomas with and without metastasis. Clin Cancer Res, 2014. 20(17): p. 4598-612.
Sproul, D., et al., Tissue of origin determines cancer-associated CpG island promoter hypermethylation patterns. Genome Biol, 2012. 13(10): p. R84.
Gregory, T.R., Synergy between sequence and size in large-scale genomics. Nat Rev Genet, 2005. 6(9): p. 699-708.
Tanaka, Y., et al., Effects of Alu elements on global nucleosome positioning in the human genome. BMC Genomics, 2010. 11:p. 309.
Kolomietz, E., et al., The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer,2002. 35(2): p. 97-112.
Price, A.L., E. Eskin, and P.A. Pevzner, Whole-genome analysis of Alu repeat elements reveals complex evolutionary history. Genome Res, 2004. 14(11): p. 2245-52.
Xiang, S., et al., Methylation status of individual CpG sites within Alu elements in the human genome and Alu hypomethylation in gastric carcinomas. BMC Cancer,2010. 10: p. 44.
邓大君, DNA 甲基化和去甲基化的研究现状及思考. 遗传, 2014. 36(05): p. 403-410.
Deng, D., Z. Liu, and Y. Du, Epigenetic alterations as cancer diagnostic, prognostic, and predictive biomarkers. Adv Genet, 2010. 71: p. 125-76.
Kelloff, G.J. and C.C. Sigman, Assessing intraepithelial neoplasia and drug safety in cancer-preventive drug development. Nat Rev Cancer, 2007. 7(7): p. 508-18.
- Beroukhim, R., et al., The landscape of somatic copy-number alteration across human cancers. Nature, 2010. 463(7283): p. 899-905.
Sun, Y., et al., Methylation of p16 CpG islands associated with malignant transformation of gastric dysplasia in a population-based study. Clin Cancer Res, 2004. 10(15): p. 5087-93.
Belinsky, S.A., et al., Promoter hypermethylation of multiple genesin sputum precedes lung cancer incidence in a high-risk cohort. Cancer Res,2006. 66(6): p. 3338-44.
Jin, Z., et al., A multicenter, double-blinded validation study of methylation biomarkers for progression prediction in Barrett’s esophagus. Cancer Res,2009. 69(10): p. 4112-5.
Cao, J., et al., Methylation of p16 CpG island associated with malignant progression of oral epithelial dysplasia: a prospective cohort study. Clin Cancer Res,2009. 15(16): p. 5178-83.
Hall, G.L., et al., p16 Promoter methylation is a potential predictor of malignant transformation in oral epithelial dysplasia. Cancer Epidemiol BiomarkersPrev, 2008. 17(8): p. 2174-9.
Lu, Z.M., et al., Nucleosomes correlate with in vivo progression pattern of de novo methylation of p16 CpG islands in human gastric carcinogenesis. PLoS One,2012. 7(4): p. e35928.
Zhou, J., et al., A 115-bp MethyLight assay for detection of p16 (CDKN2A) methylation as a diagnostic biomarker in human tissues. BMC Med Genet, 2011. 12: p. 67.
Liu, H., et al., P16 Methylation as an Early Predictor for Cancer Development From OralEpithelial Dysplasia: A Double-blind Multicentre Prospective Study.EBioMedicine, 2015. 2(5): p. 432-7.
Yin, A.A., et al., The predictive but not prognostic value of MGMT promoter methylation status in elderly glioblastoma patients: a meta-analysis. PLoS One, 2014. 9(1): p. e85102.
Ji, J., et al., MicroRNA expression, survival, and response to interferon in liver cancer. N Engl J Med, 2009. 361(15):p. 1437-47.
Ning, Z.Q., et al., Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother Pharmacol, 2012. 69(4): p. 901-9.
Liu, B., et al., Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell Metab,2012. 16(6): p. 738-50.
Chiappinelli, K.B., et al., Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses.Cell, 2015. 162(5): p. 974-86.
Zhang, B., et al., The p16-specific reactivation and inhibition of cell migration through demethylation of CpG islands by engineered transcription factors. Hum GeneTher, 2012. 23(10): p. 1071-81.
Cui, C., et al., P16-specific DNA methylation by engineered zinc finger methyltransferase inactivates gene transcription and promotes cancer metastasis. Genome Biol, 2015. 16:p. 252.
Chen, S., et al., Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell, 2015. 160(6):p. 1246-60.
推荐阅读:
ㆍ当我们在讨论表观遗传的遗传时,究竟在讨论什么?
https://mp.weixin.qq.com/s/B-Msy33k03Ld_jlcZHw_ew

若有收获,就点个赞吧
0 人点赞
