背景知识
一、目的
cgmaptools软件绘制指定基因组区域的DNA甲基化水平变化趋势
示例图1:可视化基因组上所有基因及其上下游区域的甲基化水平变化趋势
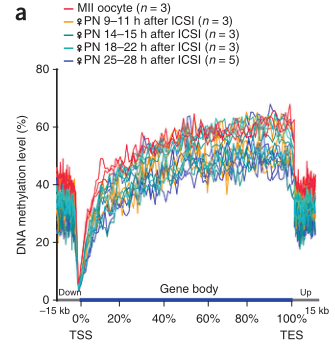
示例图2:某一个基因及其上下游区域的甲基化水平变化趋势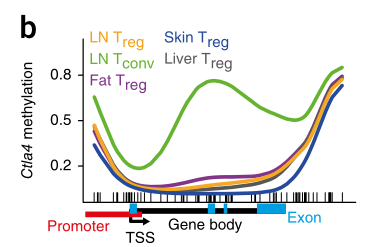
二、绘图步骤
绘制此图需要分为以下几步数据处理、运算和可视化:
- 获取cgmaptools可用的输入文件(文件格式转换)
- cgmaptools bed2fragreg
- cgmaptools mfg
- cgmaptools fragreg
三、Tips
提前理解cgmaptools bed2fragreg / mfg / fragreg这三个模块的功能,主要是他们的输入和输出文件格式;
内容较多,相比于deeptools plotProfile功能,更多的需要自己操作的地方;
参数较少但影响很大,尤其是在划分fragment长度的参数,这里需要多试几次;画这张图的最重要的部分是选择你要进行绘制的区域;
deeptools和ngsplots貌似也能画,deeptools的输入文件是bigwig格式文件,参考:https://zhuanlan.zhihu.com/p/166282791,可以使用bedGraphToBigWig小工具进行转换;
实战
以genebody的上下游区域的区域动态甲基化水平为例
Tools:shell语言
示例代码:cmd_meth_dynamics_of_targetANDflank_region.sh
version1
#!/bin/bash############################################# cgmaptools fragreg# Description: Plot methylation dynamics of target and flanking region for multiple samples,如gene body及其附件区域的甲基化水平# author: Zhiwei Lian# date: 2021.08.23# 注:cgmaptools v0.1.2的大部分模块依赖于python2,所以需要在python2的Linux环境下运行。############################################work_dir=${1}cd ${work_dir}# 1.cgmaptools bed2fragreg:Generate fragmented regions from BED file,即将指定(长度)区域划分为片段(fragment/bins)。# 建议查看一下使用说明,因为这几个参数设置只有理解了才能够设置:cgmaptools bed2fragreg --help# -n INT: 表示target regions中Number of bins to be equally split [Default:1];这里等长分割的数目是指输入的bed文件的start到end这一段区域划分为的bins数目,也就是target regions。平均划分的好处是可以避免不同target regions的不同的宽度,这也是cgmaptools fragreg要优于deeptools plotProfile的一个地方。理论上说INT越大,展示时波动越小。# -F INT_list List of region lengths in upstream of 5' end, Ex: 10,50. List is from 5'end->3'end# -T INT_list List of region lengths in downstream of 3' end, Ex: 40,20. List is from 5'end->3'end# 总的来说,需要根据数据来源(WGBS/RRBS)或者位点在这些区域的富集程度,灵活调节可视化区域的上下游长度以及每个bins的宽度(参数-n,-F,-T)# 注:输入的BED文件要求至少包含4列,即<chr> <start> <end> <strand># 首先最重要的是想好需要可视化的"target and flanking region" 的长度,并设置每个fragment/bins的宽度。# 举例:假设可视化gene body上下游的区域宽度为15kb=15000bp# 将这15kb划分的bins宽度为500,则有30个bins#500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500# 将这15kb划分的bins宽度为300,则有50个bins#300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300# genebody使用文件更新input_bed2fragreg=/mnt/MD3400/data003/lianzhiwei/01project/00RefandAnno/02_annotation/02_gene_annotation/04.UCSC-Browerand-Tool_download/hg19_GRCh37/UCSCgenes_knownGene_20201102/genebody_from_Gene/Gene_reduce_column.v2.bedcgmaptools bed2fragreg -i ${input_bed2fragreg} \-n 100 \-F 300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300 \-T 300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300 \-o cgmaptools_bed2fragreg_BinWidth300.bed# 2.使用"cgmaptools mfg" 研究片段化区域中的甲基化(methylation fragment),计算样本在上一条命令划分的每个fragment的甲基化水平,。# 该功能可用于绘制跨基因体,转座子元件和其他用户提供区域的mC分布。input_CGmap=/mnt/MD3400/data003/lianzhiwei/01project/01scRRBS/project/data/bismark_CpG_report_txt/coverage2cytosine_all_sample/scRRBS_5CellLines/bismark2cgmap/for input_filename in $(ls ${input_CGmap} | grep ".CGmap");doprefix=$(echo ${input_filename} | awk -F '.CGmap' '{print $1}')cgmaptools mfg -i ${input_CGmap}/${input_filename} -r cgmaptools_bed2fragreg_BinWidth300.bed -x CG > ${prefix}.mfgdone# tips1:最好在cgmaptools_bed2fragreg转换获得的bed文件所在文件夹下运行这一步,因为跑完这一步后容易不记得结果文件"*mfg"到底是哪个区域的甲基化水平# tips2:WGBS测序获得的位点数目达千万级别,但计算使用到的染色体区域只有一部分,为了提高运行速率,提取需要用到的那条染色体上的位点,比如我们只需要提取出 “chr14”上的CpG sitesfor input_filename in $(ls ${inut_CGmap1} | grep ".CGmap");doprefix=$(echo ${input_filename} | awk -F '.CGmap' '{print $1}')cat ${inut_CGmap1}/${input_filename} | grep ^chr14 >${prefix}.chr14.CGmapdone# 3. 合并多个样本的methylation fragment(mfg)文件为一个矩阵格式。这一步没有轮子,这是作者提供的合并代码,自己也可以写# tips1:从任意一个methylation fragment文件中获取表头并把第一个字段修改为"Sample";如文件Sample_1.mfg# tips2:提取出样本在每个fragment的平均甲基化的那行(total_ave_mC),并把样本名作为行名# tips3:这里的文件名命名采用的是sed命令替换,可以按需修改替换的字符串(head -1 H1-diagnosis.chr14.mfg | awk -F "\t" -v OFS="\t" '{$1="Sample"; print $0;}';for mfg_file in *.mfg; docat ${mfg_file} | awk -F "\t" -v OFS="\t" -v SampleName=$(echo ${mfg_file} | sed s/.mfg//g) '/total_ave_mC/{$1=SampleName;print $0}'done) > mfg_merge.xls# 4.使用cgmaptools fragreg将mfg得到的结果进行可视化cgmaptools fragreg --help# Description: Plot methylation dynamics of target and flanking region for multiple samples# -r RATIO, --ratio=RATIO:[opt] range ratio between target region and flanking region in plot. [default: 5],值越大,在结果图的横坐标,target regions占比越多,flanking region占比越少# 可视化多个样本cgmaptools fragreg -i mfg_merge.xls --ratio=2 -f pdf -o meth_fragment_merge_multi_sample.pdf# 可视化单个样本,一般不需要cgmaptools fragreg -i Sample_1.mfg -f pdf -o test_1.pdf
version2
#!/bin/bash############################################# cgmaptools fragreg# Description: Plot methylation dynamics of target and flanking region for multiple samples,如gene body及其附件区域的甲基化水平# author: Zhiwei Lian# date: 2021.08.23# 注:cgmaptools v0.1.2的大部分模块依赖于python2,所以需要在python2的Linux环境下运行。################################################################################ 传入参数#################################### 1. 运行结果和运行过程产生的文件保存路径output_path=# 2. BED格式的regions的绝对路径,精确到文件的绝对路径region_BED_path=# 3. CGmap格式的文件所在路径,精确到文件所在文件夹即可CGmap_path=#################################### step1: cgmaptools bed2fragreg: Split input region into N bins, get fragments from 5' end and 3' end.# 简单来说就是生成指定宽度的BED格式的fragment染色体坐标文件#总的来说,需要根据数据来源(WGBS/RRBS)或者位点在这些区域的富集程度,灵活调节可视化区域的上下游长度以及每个bins的宽度(参数-n,-F,-T)# 注:输入的BED文件要求至少包含4列,即<chr> <start> <end> <strand>#################################### 1.适用于输入为某个gene或者某个promoter,而非整个基因组的所有的gene或promoter。# cgmaptools bed2fragreg -i ${region_BED_path} \# -n 20 \# -F 500,500,500,500,500,500,500,500,500,500 \# -T 500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500,500 \# -o bed2fragreg_BinWidth500.bed# 2.适用于输入的BED文件为整个基因组上所有的gene,promoter,CGI等等cgmaptools bed2fragreg -i ${region_BED_path} \-n 100 \-F 300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300 \-T 300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300,300 \-o ${output_path}/cgmaptools_bed2fragreg_BinWidth300.bed#################################### step2: cgmaptools mfg计算样本在上一步获得的所有fragment的甲基化水平###################################for CGmap_filename in $(ls ${CGmap_path} | grep ".CGmap");doprefix=$(echo ${CGmap_filename} | awk -F '.CGmap' '{print $1}')cgmaptools mfg -i ${CGmap_path}/${CGmap_filename} \-r ${output_path}/cgmaptools_bed2fragreg_BinWidth300.bed \-c 5 -C 500 \-x CG > ${output_path}/${prefix}.mfgdone# default parameter:# -c Int, minimum Coverage [Default: 5]# -C Int, maximum Coverage [Default: 500]#################################### step3:合并多样本在不同fragment的甲基化水平为一个矩阵###################################first_filename=`$(ls ${output_path} | grep ".mfg" | head -1)`(head -1 ${output_path}/${first_filename} | awk -F "\t" -v OFS="\t" '{$1="Sample"; print $0;}';for mfg_file in *.mfg;docat ${mfg_file} | awk -F "\t" -v OFS="\t" -v SampleName=$(echo ${mfg_file} | sed s/.mfg//g) '/total_ave_mC/{$1=SampleName;print $0}'done) > mfg_merge.xls#################################### step4: cgmaptools fragreg将mfg得到的结果进行可视化###################################cgmaptools fragreg -i mfg_merge.xls --ratio=3 -f pdf -o meth_fragment_merge_multi_sample.pdf# -r RATIO, --ratio=RATIO:[opt] range ratio between target region and flanking region in plot. [default: 5],值越大,在结果图的横坐标,target regions占比越多,flanking region占比越少
version3
特点:输入为多个bed格式文件,以绘制多种基因组区域的这种图。
步骤1:cgmaptools bed2fragreg不在循环里面,需要提前执行,生成多个用于可视化的region(划分为fragment的BED文件)。 注意,输入的BED文件格式为:
步骤2:其它步骤使用两个循环,分别遍历所有region的BED文件和所有样本的CGmap文件,计算并出图。
步骤1:使用如下的脚本(cgmaptools_bed2fragreg.sh)将输入的bed格式的region划分为指定长度和宽度的windows。
usage:bash cgmaptools_bed2fragreg.sh
#!/bin/bash# 目的:cgmaptools bed2fragreg命令切割region及其上下游区域# 输入的BED文件所在路径# 注意:BED文件格式为:<chr> <start> <end> <strand>region_BED_path=${1}# cgmaptools bed2fragreg命令输出文件路径bed2fragreg_path=${2}# 我的参数:# 1. region这个区域均匀分割为100个Windows;# 2. regions的上下游区域长度均为5kb,均匀分割为20个Windows;for bed_file in $(ls ${region_BED_path} | grep ".bed");doregion_type=$(echo ${bed_file} | awk -F ".bed" '{print $1}')cgmaptools bed2fragreg -i ${region_BED_path}/${bed_file} \-n 100 \-F 250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250 \-T 250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250,250 \-o ${bed2fragreg_path}/${region_type}_n100ud5kbF250T250.beddone
步骤2
#!/bin/bash############################################# cgmaptools fragreg# Description: Plot methylation dynamics of target and flanking region for multiple samples,如gene body及其附件区域的甲基化水平# author: Zhiwei Lian# date: 2021.08.23# 注:cgmaptools v0.1.2的大部分模块依赖于python2,所以需要在python2的Linux环境下运行。################################################################################ 传入参数#################################### 1. 运行结果和运行过程产生的文件保存路径output_path=${1}# 2. 步骤1(cgmaptools bed2fragreg)划分bed为window的输出文件所在路径region_frag_Bed_path=${2}# 3. CGmap格式的文件所在路径,精确到文件所在文件夹即可CGmap_path=${3}#################################### 嵌套循环执行cgmaptools的两个命令#################################### 第一个循环,遍历文件夹下所有需要可视化的fragment的BED文件for frag_BED in $(ls ${region_frag_Bed_path} | grep ".bed");do# 第二个for循环,遍历文件夹下所有样本的CGmap文件for CGmap_filename in $(ls ${CGmap_path} | grep ".CGmap");do# step2: cgmaptools mfg计算多个样本在上一步获得的所有fragment的甲基化水平prefix=$(echo ${CGmap_filename} | awk -F '.CGmap' '{print $1}')cgmaptools mfg -i ${CGmap_path}/${CGmap_filename} -r ${region_frag_Bed_path}/${frag_BED} -c 5 -C 500 -x CG >${output_path}/${prefix}.mfgdone# step3:合并多样本在不同fragment的甲基化水平为一个矩阵first_filename=$(ls ${output_path} | grep ".mfg" | head -1)(head -1 ${output_path}/${first_filename} | awk -F "\t" -v OFS="\t" '{$1="Sample"; print $0;}';for mfg_file in $(ls ${output_path} | grep ".mfg");docat ${output_path}/${mfg_file} | awk -F "\t" -v OFS="\t" -v SampleName=$(echo ${mfg_file} | sed s/.mfg//g) '/total_ave_mC/{$1=SampleName;print $0}'done) >mfg_merge_${frag_BED}.xlsrm ${output_path}/*.mfg# step4: cgmaptools fragreg将mfg得到的结果进行可视化cgmaptools fragreg -i mfg_merge_${frag_BED}.xls --ratio=3 -f pdf -o meth_frag_${frag_BED}.pdfdone
不知为何,某次运行此脚本把运行的命令也添加到输出文件了,如下图: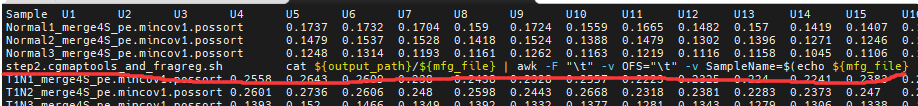
于是使用sed删除第五行:
for mfg in $(ls tmp/ | grep "mfg_mer");do sed '5d' tmp/${mfg} >${mfg}; done# 注:tmp下保存着截图出错的文件
分步骤
绘制这幅图不容易,需要分成几个步骤,代码及其说明分别如下:
step1:准备bed格式的基因及其上下游区域
cgmaptools bed2fragreg
使用说明书:
Usage: cgmaptools bed2fragreg [-i <BED>] [-n <N>] [-F <50,50,..> -T <50,..>] [-o output](aka FragRegFromBED)Description: Generate fragmented regions from BED file.Split input region into N bins, get fragments from 5' end and 3' end.Input Ex:chr1 1000 2000 +chr2 9000 8000 -Output Ex:chr1 + 940 950 1000 1200 1400 1600 1800 1850chr2 - 9060 9050 9000 8800 8600 8400 8200 8150
可选参数:
- -i FILE BED format, STDIN if omitted
- -F INT_list List of region lengths in upstream of 5’ end, Ex: 10,50. List is from 5’end->3’end
- -T INT_list List of region lengths in downstream of 3’ end, Ex: 40,20. List is from 5’end->3’end
- -n INT Number of bins to be equally split [Default:1];【这里等长分割的数目是指你输入的bed文件的start到end这一段区域划分为的bins数目,也就是target regions】
- -o OUTFILE To standard output if omitted. Compressed output if end with
实战代码
示例基因“TCL1A”,长度为8,178个碱基,只放这四列,正负链对最终出图有影响,可通过IGV等方式查看基因的正负链信息。
cat gene-TCL1A.bed# chr14 96176284 96180462cgmaptools bed2fragreg -i gene-TCL1A.bed \-n 80 \-F 100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100 \-T 100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100,100 \-o bed2fragreg_gene-TCL1A.bed
cgmaptools mfg
step2:使用”cgmaptools mfg” 研究片段化区域中的甲基化(methylation fragment)
本质:计算样本在上一条命令划分的每个fragment的甲基化水平
软件:cgmaptools mfg
使用说明(Version: 0.1.2)
Usage: cgmaptools mfg [-i <CGmap>] -r <region> [-c 5 -C 500]Description: Calculated methylation profile across fragmented regions.Contact: Guo, Weilong; guoweilong@126.comLast Update: 2017-01-20Options:-i String, CGmap file; use STDIN if not specifiedPlease use "gunzip -c <input>.gz " and pipe as input for gzipped file.chr1 G 851 CHH CC 0.1 1 10-r String, Region file, at least 4 columnsFormat: chr strand pos_1 pos_2 pos_3 ...Regions would be considered as [pos_1, pos_2), [pos_2, pos_3)Strand information will be used for distinguish sense/antisense strandEx:#chr strand U1 R1 R2 D1 Endchr1 + 600 700 800 900 950chr1 - 1600 1500 1400 1300 1250-c Int, minimum Coverage [Default: 5]-C Int, maximum Coverage [Default: 500]Sites exceed the coverage range will be discarded-x String, context [use all sites by default]string can be CG, CH, CHG, CHH, CA, CC, CT, CW-h helpOutput to STDOUT:Region_ID U1 R1 R2 D1sense_ave_mC 0.50 0.40 0.30 0.20sense_sum_mC 5.0 4.0 3.0 2.0sense_sum_NO 10 10 10 10anti_ave_mC 0.40 0.20 0.10 NaNanti_sum_mC 8.0 4.0 2.0 0.0anti_sum_NO 20 20 20 0total_ave_mC 0.43 0.27 0.17 0.2total_sum_mC 13.0 8.0 5.0 2.0total_sum_NO 30 30 30 10
批量运行代码
inut_CGmap1=/mnt/MD3400/data003/lianzhiwei/01project/03WGBS_ALL/result/Meth_Level_dynamics/cgmaptools_plot/input_bismark2CGmap# WGBS测序获得的位点数目达千万级别,但计算使用到的染色体区域只有一部分,为了提高运行速率,提取需要用到的那条染色体上的位点for input_filename in $(ls ${inut_CGmap1} | grep ".CGmap");doprefix=$(echo ${input_filename} | awk -F '.CGmap' '{print $1}')cat ${inut_CGmap1}/${input_filename} | grep chr14 >${prefix}.TCL1A.CGmapdoneinut_CGmap2=/mnt/MD3400/data003/lianzhiwei/01project/03WGBS_ALL/result/Meth_Level_dynamics/cgmaptools_plot/gene_MethLevel_20210503/gene-TCL1A/filtered_CGmapfor input_filename in $(ls ${inut_CGmap2} | grep ".CGmap");doprefix=$(echo ${input_filename} | awk -F '.CGmap' '{print $1}')cgmaptools mfg -i ${inut_CGmap2}/${input_filename} \-r /mnt/MD3400/data003/lianzhiwei/01project/03WGBS_ALL/result/Meth_Level_dynamics/cgmaptools_plot/gene_MethLevel_20210503/gene-TCL1A/bed2fragreg_gene-TCL1A.bed \-c 5 \-C 500 \-x CG > ${prefix}.mfgdone# default parameter:# -c Int, minimum Coverage [Default: 5]# -C Int, maximum Coverage [Default: 500]# Sites exceed the coverage range will be discarded# -x String, context [use all sites by default]# string can be CG, CH, CHG, CHH, CA, CC, CT, CW
merge mfg
step3:合并多个样本的methylation fragment(mfg)文件为一个矩阵格式。
这一步没有轮子,这是作者提供的合并代码,自己也可以写
# tips1:从任意一个methylation fragment文件中获取表头并把第一个字段修改为”Sample”;如文件Sample_1.mfg
# tips2:提取出样本在每个fragment的平均甲基化的那行(total_ave_mC),并把样本名作为行名
# tips3:这里的文件名命名采用的是sed命令替换,可以按需修改替换的字符串
(head -1 H1-diagnosis.chr14.mfg | awk -F "\t" -v OFS="\t" '{$1="Sample"; print $0;}';for mfg_file in *.mfg; docat ${mfg_file} | awk -F "\t" -v OFS="\t" -v SampleName=$(echo ${mfg_file} | sed s/.chr14.mfg//g) '/total_ave_mC/{$1=SampleName;print $0}'done) > mfg_merge.xls
cgmaptools fragreg
step4:使用cgmaptools fragreg将mfg得到的结果进行可视化
cgmaptools fragreg —help
# Description: Plot methylation dynamics of target and flanking region for multiple samples
# -r RATIO, —ratio=RATIO:[opt] range ratio between target region and flanking region in plot. [default: 5],值越大,在结果图的横坐标,target regions占比越多,flanking region占比越少
# 可视化多个样本cgmaptools fragreg -i mfg_merge.xls --ratio=2 -f pdf -o meth_fragment_merge_multi_sample.pdf# 可视化单个样本,一般不需要cgmaptools fragreg -i Sample_1.mfg -f pdf -o test_1.pdf
绘制目标图形的几个步骤已经完成,接下来需要做的是修图。
报错合集
- 绘图需要的R包没安装
cgmaptools fragreg需要调用当前环境下的R来绘图,如果当前环境没有绘图需要的R包,如这里报错显示R包optparse没安装,只能先安装好R包,再运行cgmaptools fragreg绘图。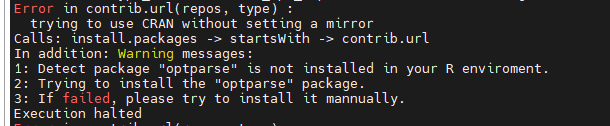
但是如果前面几步完成了,用于绘图的矩阵文件也就有了(mfg_merge.xls),下一步是使用cgmaptools fragreg的绘图源代码进行绘图。
ps:因为cgmaptools fragreg 命令绘图结果不会对图中的折线按照分组进行上色,而是每一个样本一条不同颜色的曲线,如果输入样本里面分为不同组别,一组内有多个样本,那么cgmaptools fragreg 命令出图结果就不可用了。如下图: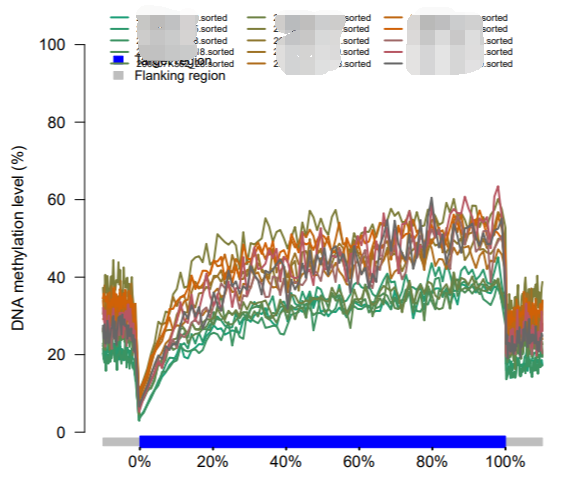
遇到这种情况,就要重新自己使用矩阵绘图。
问题:软件原始代码绘制的图分组上色可能不合理,需要手动调整cgmaptools在GitHub上的绘制此图的R代码。
cgmaptools fragreg这个shell命令行下使用的绘图命令实际上依赖于,其R源代码可获取于可获取于:https://github.com/guoweilong/cgmaptools/blob/master/src/mCFragRegView.R
cgmaptools fragreg的源代码经过修改后可适用于分组显示的R代码:
修改cgmaptools绘图函数源码_input_methylationFragment矩阵.R
目的:修改cgmaptools绘图函数源码_input_methylationFragment矩阵
后期搭配AI、PS等工具的使用即可获得一张可发表的图形。
优化与修图
- 修图是什么意思呢?
理想很丰满,我们都希望绘制的图形一次就能达到满意效果,但是现实上,你的数据和参数往往给你一个惊喜。
比如下图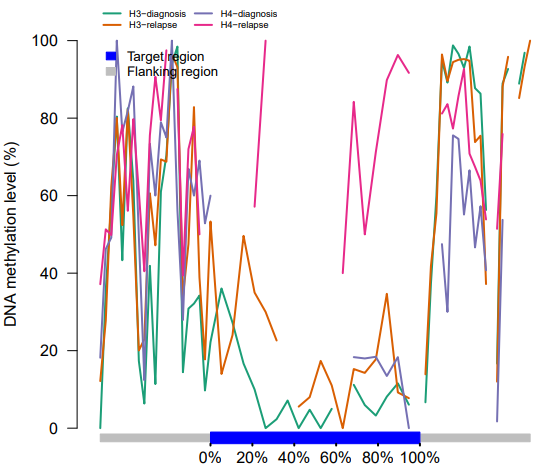
为何每个样本的甲基化水平没有连成线?原因可能有二:
一是样本检测到的位点数目太少,导致划分bins的区域没有位点所以该区域为NA,线就连不上;
二是划分bins时bins的宽度太窄,导致很多bins没有位点所以该区域为NA,线就连不上;
然后解决办法是,多测试参数,放宽或缩小参数。
- 优化
有时间按照这种格式绘图,方法是将cgmaptools输出的mfg文件拼接起来,再使用cgmaptools fragreg命令绘图

若有收获,就点个赞吧
0 人点赞
