deeptools可视化基因组区域的甲基化水平需要使用到3个命令:computeMatrix,plotProfile,plotHeatmap;
官网说明书:
deeptools 说明文档
deeptools的GitHub
deeptools computeMatrix 说明文档
deeptools plotHeatmap 说明文档
deeptools tutorial
CpG methylation around murine transcription start sites in two different cell types
除了组蛋白尾部的甲基化外,胞嘧啶也可以被甲基化(有关 CpG 甲基化的更多信息,请阅读此处)。 在哺乳动物基因组中,大多数 CpG 被甲基化,除非它们位于需要保持未甲基化以允许完全转录活动的基因启动子中。
在以下热图中,我们使用了主要在 ES 细胞中表达的基因,并检查了其转录起始位点(TSS)周围甲基化胞嘧啶的百分比。 蓝色信号表明发现很少的甲基化胞嘧啶。 当您比较 ES 细胞和神经元祖 (NP) 细胞之间的 CpG 甲基化信号时,您可以看到大多数基因保持未甲基化,但 TSS 周围的 CpG 甲基化的一般量增加,如更强的红色信号和 摘要图中 CpG 甲基化信号略有升高。 这支持了存储在 BED 文件中的基因确实倾向于在 ES 中比在 NP 细胞中表达更多的观点。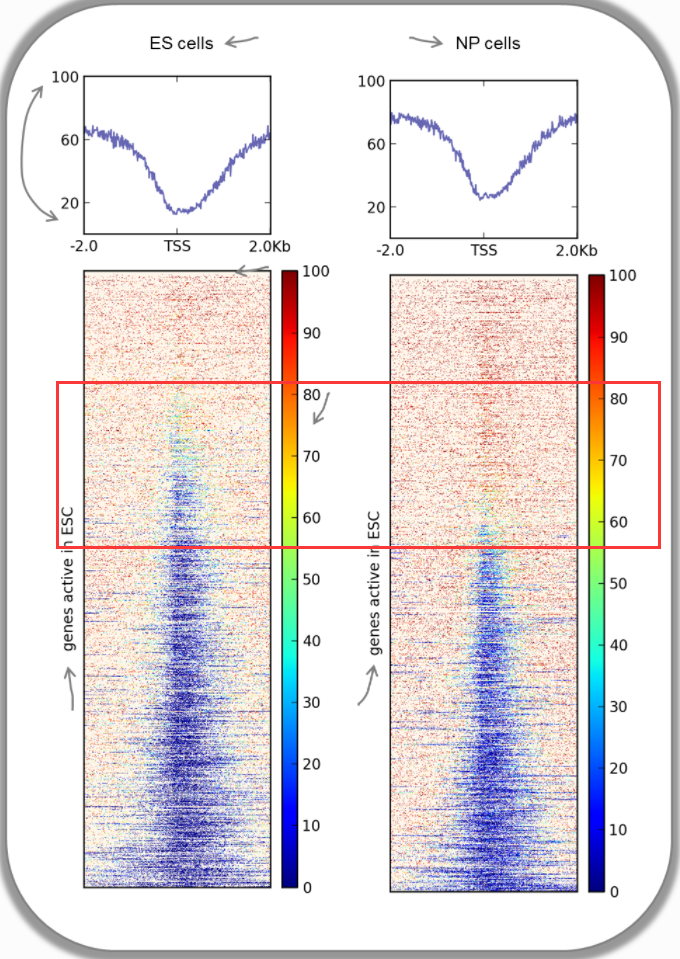
| Fast Facts | |
|---|---|
| computeMatrix mode | reference-point |
| regions files | BED file mouse genes expressed in ES cells |
| signal file | bigWig files with fraction of methylated cytosins (from Stadler et al., 2011) |
| heatmap cosmetics | color scheme, labels, titles, color for missing data was set to customized color, y-axis of profiles were changed, heatmap height |
以下是deeptools给出的代码,对ES和NP细胞的bigWig文件均运行一次下面的计算和绘图代码。这里的plotHeatmap命令的参数与其默认参数有一点不同,是专门适用于甲基化数据可视化。
第一步:计算修饰信号值(computeMatrix),这里我们的修饰信号就是位点的甲基化水平
$ computeMatrix reference-point \-S GSE30202_ES_CpGmeth.bw \-R activeGenes_ESConly.bed \--referencePoint TSS \-a 2000 -b 2000 \-out matrix_Genes_ES_CpGmeth.tab.gz
更多参数的computeMatrix使用方法,来自:多样本修饰信号线图(computeMatrix-ggplot2)
# 输入,需要计算的bed3文件,即包含chr,start,end三列,tab分隔的文件input_bed3=<path to bed3 file> # 输入,bigwig文件路径input_bw=<path to bigwig file> # 输出,信号值矩阵(关键结果,用于后续画图)output_matrix=./output_denisty.gz # 输出,过滤信号值为0的bed文件output_bed=./output_bed # deptools的computMatrix计算信号矩阵computeMatrix reference-point \# 计算模式,center,TSS or TES,即计算什么位置的信号--referencePoint center \# 区间的宽度-bs 20 \# -b 扩展到上游的距离, -a 扩展到下游的距离-b 50000 -a 50000 \# 输入计算的bed文件-R ${input_bed3} \# 输入bw文件-S ${input_bw} \# 是否去除信号为0的区域--skipZeros \# 输出信号矩阵-o ${output_matrix} \# 输出过滤后的bed文件--outFileSortedRegions ${output_bed} \# 线程数量-p 5
第二步:绘制修饰信号的热图(plotHeatmap)
$ plotHeatmap \-m matrix_Genes_ES_CpGmeth.tab.gz \-out hm_activeESCGenes_CpG_ES_indSort.png \--colorMap jet \--missingDataColor "#FFF6EB" \--heatmapHeight 15 \--yMin 0 --yMax 100 \--plotTitle 'ES cells' \--regionsLabel 'genes active in ESC'
准备输入文件
1. bigWig文件
一、原始文件是bedGraph格式文件
目的:Convert a bedGraph file to bigWig format
工具:UCSC的文件格式转换小工具之bedGraphToBigWig,地址:https://github.com/ENCODE-DCC/kentutils_v385_bin_bulkrna(可执行的文件)。
bedGraphToBigWig Usage:bedGraphToBigWig <in.bedGraph> <chrom.sizes> <out.bw>
参数说明:
:bedGraph文件格式的CpG位点信息,可从BS-seq常用比对工具bismark的”bismark_methylation_exactor”功能获取; :指定输出的BigWig 格式文件名; :染色体编号及其大小(碱基个数); ```bash chr1 249250621 chr2 243199373 chr3 198022430 chr4 191154276
“chrom.sizes”文件获取方法: 1). 利用 samtools 或者 fetchChromSizes 等工具从基因组序列文件(fasta格式)计算获得 samtools faidx hg38.fa
2). 相关数据库下载,如UCSC:hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/
须知:1. 输入的bedGraph文件需要先按照染色体排序,可使用sort进行排序:`sort -k1,1 -k2,2n unsorted.bedGraph > sorted.bedGraph`;注意在处理是需要跳过bedGraph文件的首行,因为bedGraph文件首行默认为表头,一经排序后就会在文件最后一行,不容易发现,如果直接使用bedGraphToBigWig命令,就会报错:`Expecting at least 3 words line 2313085[文件的行数,即最后一行] of merge_MGC803_2S.sorted.bedGraph got 2`。2. bedGraphToBigWig 输入的bedgraph文件需要按照染色体坐标排序且不允许存在染色体坐标重叠。否则会报错如下:3. 注意输入的bedGraph是压缩(cat)还是非压缩格式(zcat)。```bash#!/bin/bash#-------------------------------># Aim: 将bedGraph格式文件转换为bigwig格式文件,用于下游分析,如deeptools# input file: bedGraph file# Tools: shell, bedGraphToBigWig#-------------------------------># 传入参数1:bedGraph文件所在路径inputfile_path=${1}# bedGraphToBigWig安装位置:bedGraphToBigWig=1.bedGraphToBigWig/bedGraphToBigWig# Usage:bedGraphToBigWig <in.bedGraph> <chrom.sizes> <out.bw># chrom.sizes, GRCh38/hg19 genome versionchrom_sizes=ref_genome_fa/chrom_sizes/hg38.chrom.sizesfor bedGraph in $(ls ${inputfile_path} | grep ".bedGraph");doprefix=$(echo ${bedGraph} | awk -F ".bedGraph" '{print $1}')cat ${inputfile_path}/${bedGraph} | awk 'NR>1{print}'| grep -vE "^chrUn|^chr.*alt|^chr.*fix|^chr.*random|^chrKI|^chrGL" | sort -k1,1 -k2,2n >${prefix}.sorted.bedGraph${bedGraphToBigWig} ${prefix}.sorted.bedGraph ${chrom_sizes} ${prefix}.bw# rm ${prefix}.sorted.bedGraphdoneecho "The work is complete: $(date +%Y-%m-%d\ %H:%M:%S)"
二、如果原始文件不是bedGraph格式的文件,比如基因组区域的注释结果(即每个/每种基因组区域的甲基化水平),则需要根据文件的列顺序,提取出与bedGraph格式的chr, start, end, methylation_level这四列,,然后和上面一样,排序后再使用bedGraphToBigWig转换为bigwig即可。
注意是否输入文件是否有表头,如果有则需要跳过表头。
以下是我对自己获得的的基因组区域注释结果文件进行信息提取的shell代码。
#!/bin/bash#-------------------------------># Aim: 将BS-seq数据注释到基因组区域的结果转换为bigwig格式文件,用于下游分析,如deeptools# input file: 基因组区域注释结果# Tools: shell, bedGraphToBigWig#-------------------------------># 传入参数1:基因组区域注释结果所在路径inputfile_path=${1}# bedGraphToBigWig安装位置:bedGraphToBigWig=cmd_Genomic_Browser_Visual/1.bedGraphToBigWig/bedGraphToBigWig# Usage:bedGraphToBigWig <in.bedGraph> <chrom.sizes> <out.bw># chrom.sizes, GRCh38/hg19 genome versionchrom_sizes=ref_genome_fa/chrom_sizes/hg38.chrom.sizesfor txt in $(ls ${inputfile_path} | grep ".txt");doprefix=$(echo ${txt} | awk -F ".txt" '{print $1}')# 使用grep命令根据字符匹配原则过滤掉非参考染色体cat ${inputfile_path}/${txt} | grep -vE "^chrUn|^chr.*alt|^chr.*fix|^chr.*random|^chrKI|^chrGL|^chrJH" | awk -F "\t" -v OFS="\t" '{if($3>=5) print $4,$5,$6,$1}' | sort -k1,1 -k2,2n >${prefix}.sorted.bedGraph${bedGraphToBigWig} ${prefix}.sorted.bedGraph ${chrom_sizes} ${prefix}.bwrm ${prefix}.bedGraphdoneecho "The work is complete: $(date +%Y-%m-%d\ %H:%M:%S)"
2. bed-基因组区域文件
一般如gene body、TSS、promoter等基因组区域是比较常用于deeptools可视化的对象,这些基因组区域的坐标可以从UCSC Table Browser下载。
computeMatrix + plotHeatmap 绘图
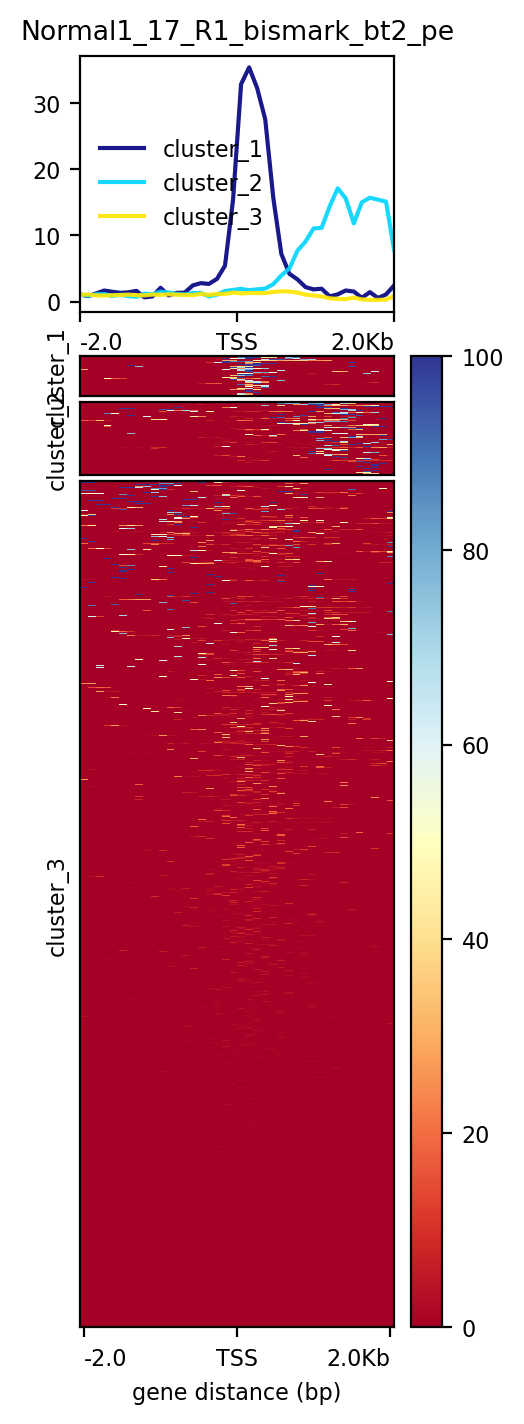
#!/bin/bashactiveGenes=ref_genome_fa/Mus_musculus/BED_GenomicRegions/ChrFilter/Gene_proteinCode.filtered.bedbwfile=Normal1_17_R1_bismark_bt2_pe.bwmark=Normal1_17computeMatrix reference-point \-S ${bwfile} \-R ${activeGenes} \--binSize 100 \--skipZeros \-p 10 \--referencePoint TSS \-a 2000 -b 2000 \-out matrix_Genes_${mark}_CpGmeth.tab.gzplotHeatmap \-m matrix_Genes_${mark}_CpGmeth.tab.gz \-out mm_${mark}_active_CGenes_CpG.png \--whatToShow 'plot, heatmap and colorbar' \--colorMap jet \--missingDataColor "#FFF6EB" \--heatmapHeight 15 \--yMin 0 --yMax 100 \--regionsLabel 'Protein coding gene'# 可选添加的参数:# --zMin 0 --zMax 100 \# --kmeans 4 \
实际结果如下:因为NA太多,导致热图区域都是NA指定的颜色(#FFF6EB)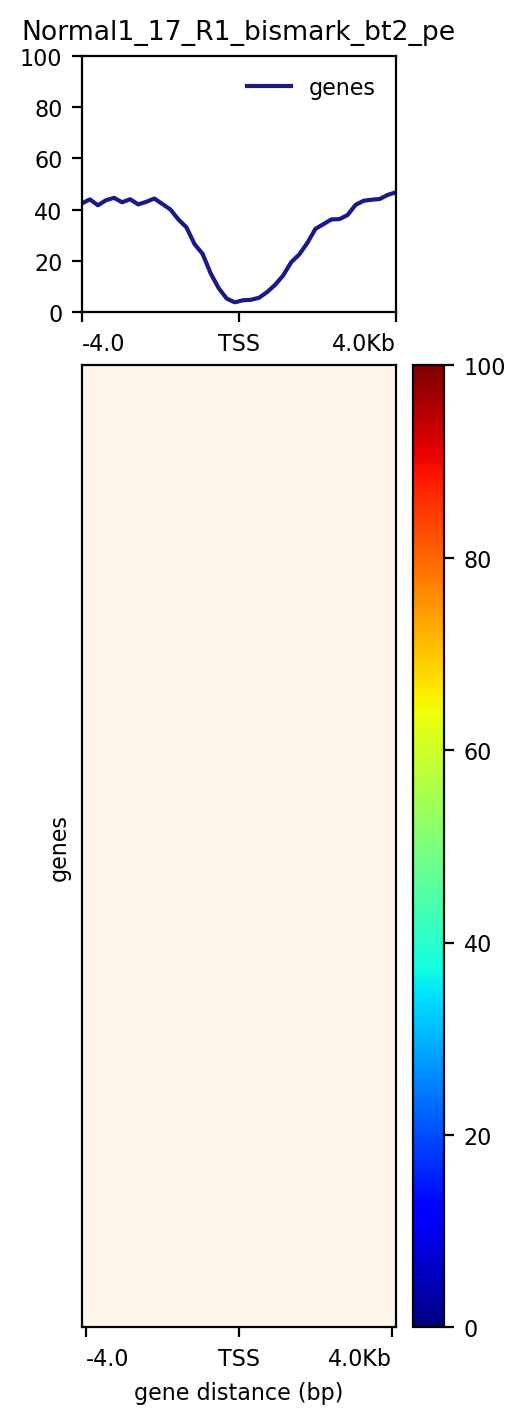
- 测试过plotHeatmap 命令将NAN的颜色设置为白色,结果热图区是“白板”;
- 测试过computeMatrix 命令添加参数
--skipZeros,出来的图形和上面的完全一样,没有变化,因为这个参数是去除的是整行都为NAN;
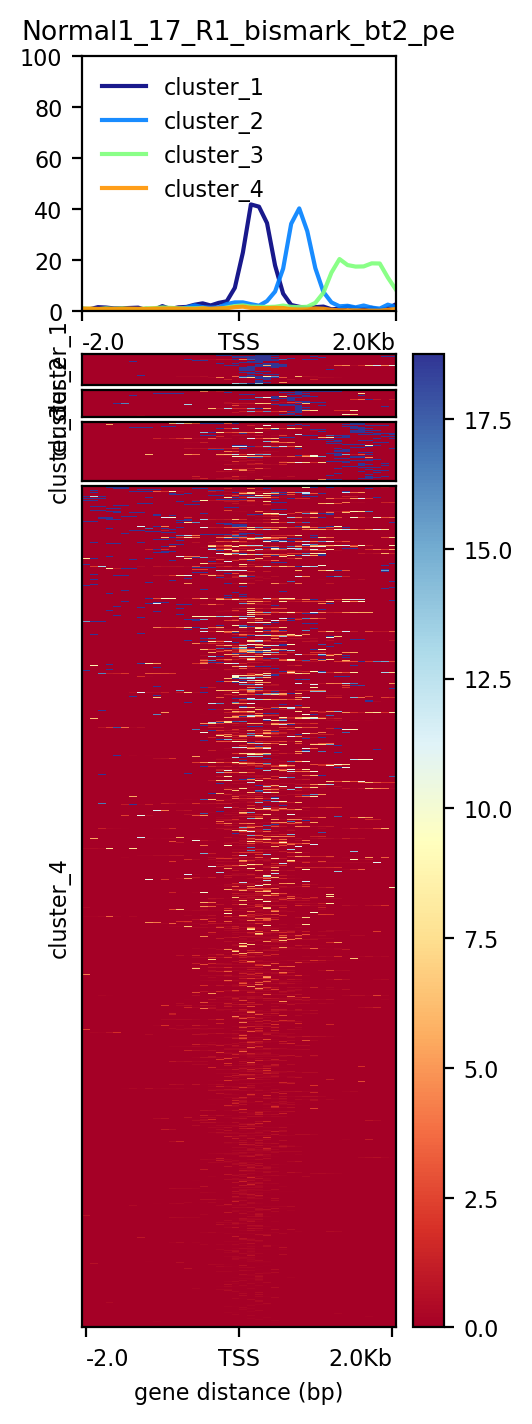
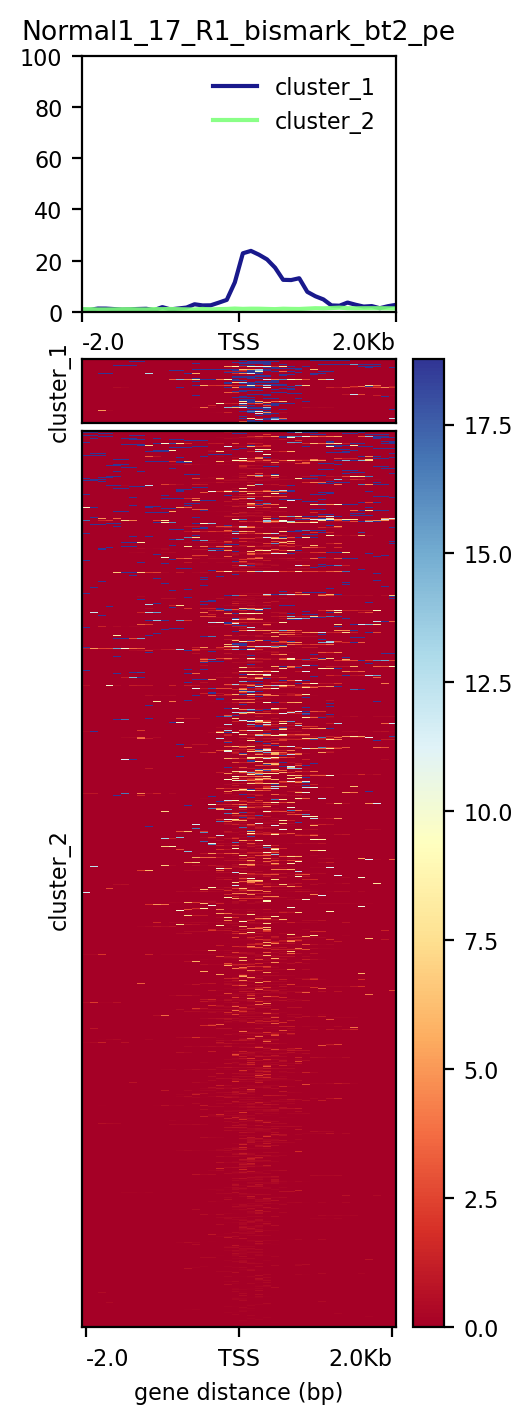
修图1:R语言过滤行
将矩阵文件读入到R,过滤掉NAN值较多的行(输入的bed文件中的行),然后再进行绘图。
setwd("D:/lianzhiwei/lzw/生化组工作/甲基化/01.潘组RRBS/02.下游分析/result/06.区域甲基化水平/deeptools_heatmap/小鼠肝及肝癌样本/2st/3st_refPoint")mat <- read.table(file="test.tab", skip=1)dim(mat)#因为deeptools computeMatrix输出的矩阵首行为命令参数记录,所以需要跳过,保存文件后需要从原文件复制原文件的首行到生成的文件中,且需要修改首行的group_boundaries":[0,矩阵行数-1]"############################# 过滤缺失值较多的行############################# 提取甲基化水平矩阵filtered_mat <- mat[, c(7:ncol(mat))]dim(filtered_mat)# 过滤一部分缺失值较多的行keep_row <- c()for (i in 1:nrow(filtered_mat)) {if (table(filtered_mat[i,] == "NaN")[[2]] < ncol(filtered_mat)/2) {keep_row <- append(keep_row, i)}}mat <- mat[keep_row,]dim(mat)mat <- as.data.frame(mat)write.table(mat, file = "mat.tab", quote = F, sep = "\t", col.names = F, row.names = F)# 将[0,100] scale to [-50,50]############################# 将非NA的值总体减去50,使得0用于存储NA值mode(filtered_mat)filtered_mat <- as.data.frame(filtered_mat)sub_mat <- filtered_mat-50sub_mat <- sub_mat[keep_row,]dim(sub_mat)write.table(sub_mat, file = "mat_subtract50.tab", quote = F, sep = "\t", col.names = F, row.names = F)# 注意,使用R保存有缺失值的矩阵时,缺失值记录为"NA",在deeptools plotHeatmap里面缺失值必须为"nan",# 所以这个文件还需要使用shell语言将"NA"完全替换为"nan"# cat mat.tab | sed 's/NA/nan/g' | gzip >mat.tab.gz############################# 尝试:pheatmap使用过滤缺失值较多的行后的矩阵进行热图绘制############################filtered_mat <- filtered_mat[keep_row,]# 没有有意义的行名pheatmap::pheatmap(filtered_mat,cluster_cols = F)
修图2:shell语言过滤行
重点:AWK对行进行字符出现次数统计并根据字符频率筛选符合条件的行。
注意AWK的for循环和if判断语句的位置,又是踩坑的一天
cat test.tab | awk '{for(i=1;i<=NF;i++){if($i=="nan") a++} {if(a<=NF/2) print};a=0}' >mat.tabvim mat.tab #修改首行的group_coundaries:[0,N]部分,其中N= 矩阵文件的行数-1。gzip mat.tab# AWK踩坑记录# cat head.tab | awk '{for(i=1;i<=NF;i++){if($i=="nan") a++} print a}' #遍历每一行,但记录NA的变量没有在每行遍历前重置为零# cat head.tab | awk '{for(i=1;i<=NF;i++){if($i=="nan") a++}}END{print a}' #已经遍历所有行,输出的a是所有行累加的a
左图是没有进行聚类,右图是使用--kmeans进行聚类了,但是由于plotHeatmap会默认把缺失值填补为0,这与DNA甲基化中的非甲基化的甲基化水平值(也是零)冲突,所以会导致整个热图都是0的色度条指定的颜色,如右图的蓝色。
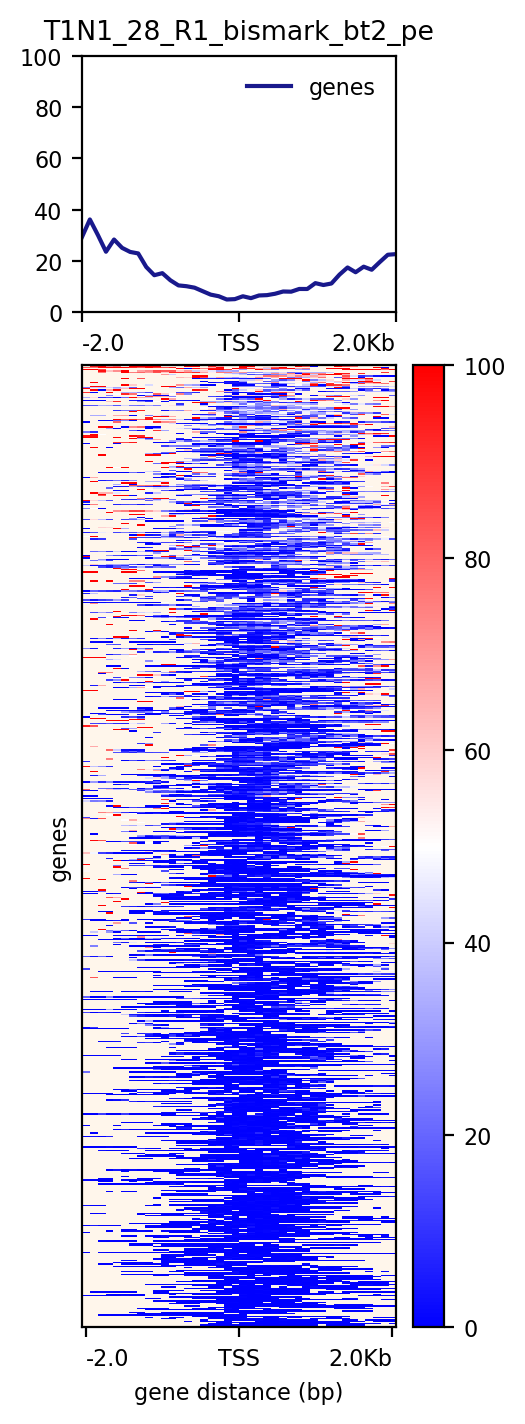
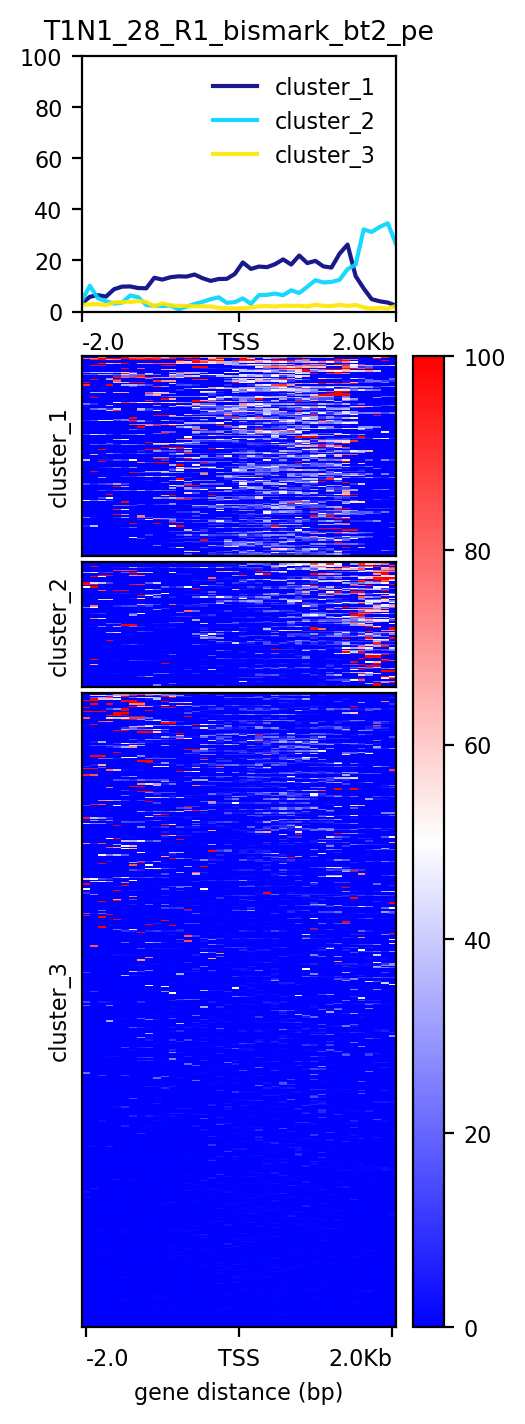
实测,即使指定--missingDataColor,出来的图还是和右图一样,缺失值的颜色还是和零一样。
plotHeatmap -m mat.tab.gz -out test5.png —colorList ‘blue,white,red’ —missingDataColor “white” —heatmapHeight 15 —zMin 0 —zMax 100 —yMin 0 —yMax 100 —kmeans 3
(optional)不筛选行,仅仅将甲基化水平从[0,100]转换为[-50,50],因为上面描述的原因,所以需要进行此类型的转换。
zcat rmHalfNA_mat.tab.gz | awk -F "\t" -v OFS="\t" '{for(i=7;i<=NF;i++){if($i!="nan") $i=$i-50}print}' | gzip >rmHalfNA_mat_sub50.tab.gzplotHeatmap -m rmHalfNA_mat_sub50.tab.gz -out test2.png --colorList 'blue,white,red' --heatmapHeight 15 --missingDataColor "white" --zMin -50 --zMax 50 --kmeans 3
(optional)筛选行的同时,将甲基化水平从[0,100]转换为[-50,50]
zcat rmHalfNA_mat.tab.gz | awk -F "\t" -v OFS="\t" '{for(i=7;i<=NF;i++){if($i=="nan") a++; else $i=$i-50}{if(a<=NF/2) print};a=0}' >rmHalfNA_sub50_mat.tabwc -l rmHalfNA_sub50_mat.tabvim rmHalfNA_sub50_mat.tab #修改首行的group_coundaries:[0,N]部分,其中N= 矩阵文件的行数-1。gzip rmHalfNA_sub50_mat.tabplotHeatmap -m rmHalfNA_sub50_mat.tab.gz -out test2.png --colorList 'blue,white,red' --heatmapHeight 15 --missingDataColor "white" --zMin -50 --zMax 50 --kmeans 3
左图:肿瘤样本;右图:健康对照样本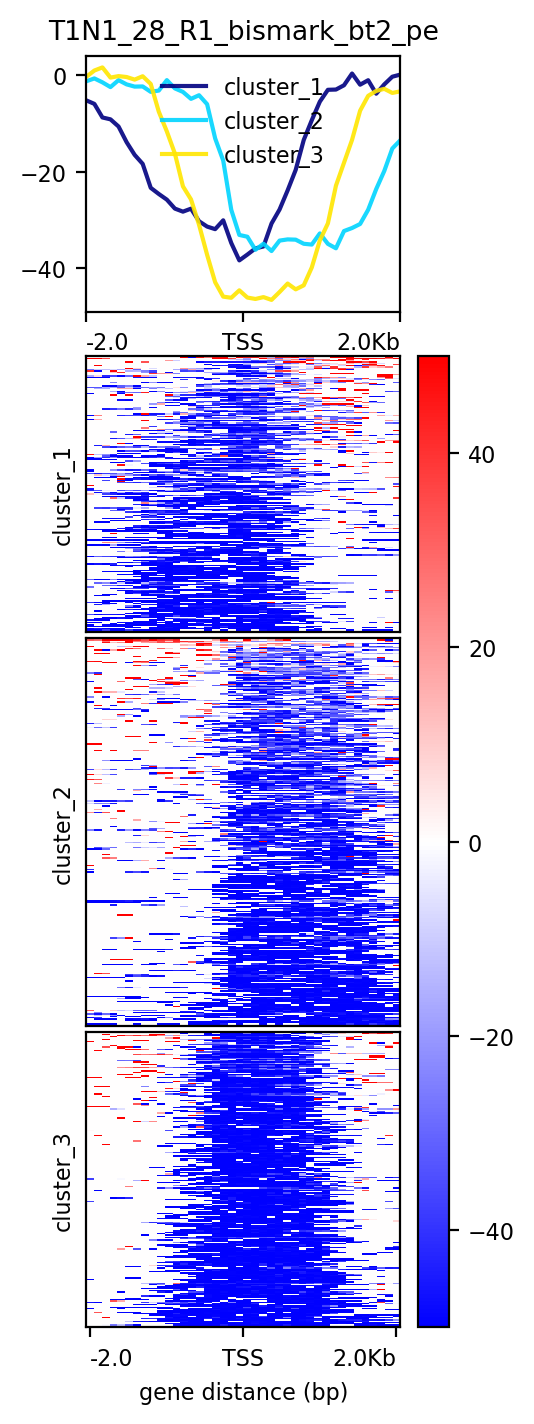
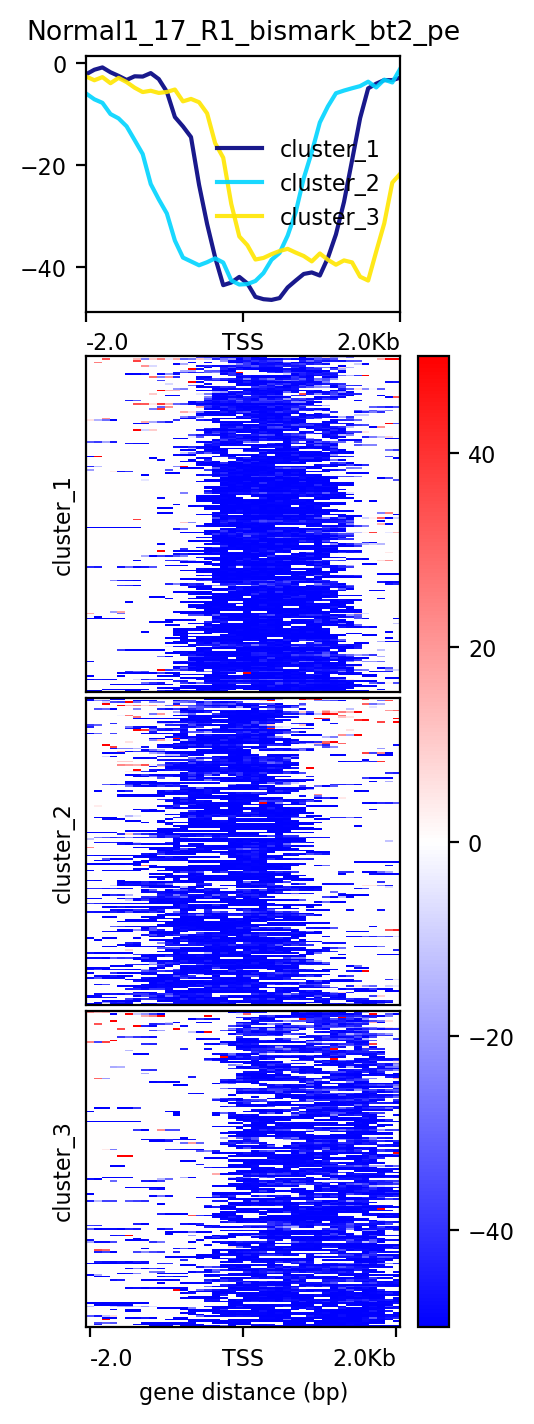
可能是computeMatrix命令的--binSize 100和-a 2000 -b 2000设置不太合理,或者此批数据来源于RRBS的原因,热图显示TSS上下游2kb区域均为低甲基化,调整参数再试一下。
实战:多个样本
#!/bin/bashactiveGenes=Gene-proteinCodeGene.bedsample1=K562.bwsample2=GM12878.bwsample3=MDA-MB-231.bwsample4=MGC803.bwsample5=293T.bwsample6=KG-1a_NR.bwcomputeMatrix reference-point \-S ${sample1} ${sample2} ${sample3} ${sample4} ${sample5} ${sample6}\-R ${activeGenes} \--binSize 100 \--skipZeros \-p 10 \--referencePoint TSS \-a 2000 -b 2000 \-out matrix_Genes_CpGmeth.tab.gz# 可视化图一:把所有的bins作为绘图输入plotHeatmap \-m matrix_Genes_CpGmeth.tab.gz \-out active_CodeGene_CpG.png \--colorMap jet \--missingDataColor "#FFF6EB" \--heatmapHeight 15 \--yMin 0 --yMax 100 \--zMin 0 --zMax 100 \--kmeans 4# (optional)可视化图二:对computeMatrix命令计算出来的结果进行筛选后再绘图,一般是去除NA较多的行# 筛选行的同时,将甲基化水平从[0,100]转换为[-50,50]zcat matrix_Genes_CpGmeth.tab.gz | awk -F "\t" -v OFS="\t" '{for(i=7;i<=NF;i++){if($i=="nan") a++; else $i=$i-50}{if(a<=NF/2) print};a=0}' >rmHalfNA_sub50_mat.tabvim rmHalfNA_sub50_mat.tab #修改首行的group_coundaries:[0,N]部分,其中N= 矩阵文件的行数-1。gzip rmHalfNA_sub50_mat.tabplotHeatmap -m rmHalfNA_mat_sub50.tab.gz -out test.png --colorList 'blue,white,red' --heatmapHeight 15 --missingDataColor "white" --zMin -50 --zMax 50 --kmeans 4
踩坑记录
踩坑1
凡是对computeMatrix命令输出的矩阵文件进行过修改的都会报一些错误。
比如:行最大值与矩阵形状不匹配???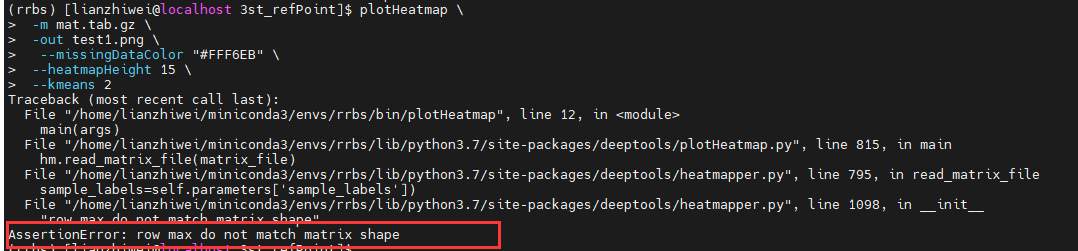
原因
修改了矩阵文件的行数后,表头文件如果没有修改就会报错,如
$ zcat matrix.tab.gz | wc -l #14485行
其表头为
解决办法:
表头与文件的行列数一致的话,绘图就不会报这种错误。所以修改矩阵文件的表头group_coundaries:[0,N],其中N= 当前矩阵文件的行数-1。
FAQ
plotProfile绘制的热图的每一行对应于输入的regions文件的一行;但如果在
computeMatrix命令使用了--skipZeros参数,则热图中的行数可能会少于regions文件的行。注意bigWig文件中的超大值,如Chip-seq中的某个peak如果非常高,可能会影响到其它peak的展示,但CpG sites不存在这种情况,因为甲基化水平的区间在[0, 100]。
非常需要注意的是,输入文件为甲基化测序数据时,
computeMatrix命令千万不要使用--missingDataAsZero,这会将所有非 CpG 和未覆盖的位置转换为看起来像它们具有 0 甲基化,这将极大地扭曲结果。参考deeptools的GitHub上的问题。
同时,使用plotHeatmap命令绘制热图时,如果矩阵内存在NA(一般都会存在),为了能进行聚类(因为有个--kmeans参数),deeptools会默认使用0来填充这个缺失值,并打印警告:Warning* For clustering nan values have to be replaced by zeros,这对于Chip-seq数据来说没啥影响,但是对于输入为CpG sites 的甲基化水平的数据来说,这会引起歧义,因为在非甲基化的甲基化水平值也为0,如果把缺失值填充为0,就相当于引入非甲基化的regions,从而使得热图总体甲基化水平偏低。
在绘制热图之前对矩阵进行额外的处理,例如去除异常值。 我们发现这在大多数情况下都是有益的。 您可以分别通过手动设置—zMax 和/或--zMin 来覆盖它。<br />参数—whatToShow`:默认情况下,在热图顶部包含一个摘要或概要图和一个热图颜色栏。其他选项有:plot and heatmap, heatmap only, heatmap and colorbar,以及默认的plot, heatmap and colorbar。
可能出现的问题1:输入的bed文件存在重叠的基因组区域,如下
$ cat testBed.bed
chr1 10 20 region1
chr1 7 15 region2
chr1 18 29 region3
chr1 35 40 region4
chr1 10 20 region1Duplicate
computeMatrix如何处理重叠的基因组区域,官方给出的解决办法如上链接。
- plotHeatmap命令如何指定绘图使用的颜色?
首先参考plotHeatmap命令的说明文档,绘制热图的颜色主要有--colorList和--colorMap两个参数

而DNA甲基化数据一般使用红色代表甲基化,蓝色代表非甲基化,所以我会选择使用--colorList参数直接指定颜色。
plotHeatmap -m rmHalfNA_mat.tab.gz -out test3.png --colorList 'blue,white,red' --missingDataColor "#FFF6EB" --heatmapHeight 15 --zMin 0 --zMax 100 --yMin 0 --yMax 100
对于CpG 位点检测数量有限的DNA甲基化测序技术,如单细胞样本,RRBS等,由于其位点稀疏性,有很大一部分基因组区域可能没有位点覆盖,所以使用computeMatrix 计算区域内的甲基化水平时,很大一部分都为NAN,即没有位点覆盖。因此绘制此类型图适合WGBS或者将多个相同类型或技术重复的样本合并后再进行绘图。
(未确定)如果输入文件是BedGraph格式,需要将它的所有
start或end的坐标增加1。因为BedGraph格式文件的坐标系统是0-based。原话:BedGraph files are 0-based, half open, so the first entry should have ended at 1. 在提问的末尾。
自由性更大的热图绘制方法
方法一:computeMatrix(shell)+ggplot2(R语言)绘制多个样本的表观修饰信号图
参考知乎的教程:多样本修饰信号线图(computeMatrix-ggplot2)
注:上面computeMatrix使用方法里面也引用了这个链接
方法二:R包—EnrichedHeatmap
http://bioconductor.org/packages/devel/bioc/vignettes/EnrichedHeatmap/inst/doc/roadmap.html

若有收获,就点个赞吧
0 人点赞
