RnBeads官网:https://rnbeads.org/
RnBeads已经在2019年升级到2.0版本,文章发表在 Genome Biology,地址:https://doi.org/10.1186/s13059-019-1664-9
RnBeads是什么
RnBeads是一个强大的R包,用于综合分析通过任何提供单CpG分辨率的实验方案获得的DNA甲基化数据,支持的测定包括Infinium和EPIC芯片以及亚硫酸氢盐测序方案,以及MeDIP-seq和MBD-seq。RnBeads实施的分析工作流程比现有工具要全面得多。它以高度注释和可读的超文本报告形式记录其结果,并且可以扩展为大样本量,这些样本量已成为人类群体DNA甲基化分析的标准。
支持各种DNA甲基化测定和输入格式。管道实现了最新的规范化技术。可以进行实验质量控制,并可以识别异常值和混合样品。该软件包提供了用于CpG和样本过滤的灵活方法。根据样本注释,可以识别批次效应和表型协变量。分析DNA甲基化分布,并量化甲基化谱中的族间和组内变异性。此外,可以表征样品组之间的差异甲基化。该分析基于单个CpG以及预定义或自定义的基因组区域。最后,甲基化数据可以多种格式导出,包括基因组浏览器视图。综合的,生成高度解释性的报告,其中包含方法说明,出版物等级图表和数据表。它们的HTML格式有助于轻松跟踪和比较分析结果,并与合作伙伴交换结果。由于其模块化的概念,首次使用的用户和专家都可以根据他们的个人需求方便地执行分析。可以通过仅指定几个参数并执行主命令来调用一次全面的分析运行。或者,用户可以单独执行管道的步骤。初次使用的用户和专家都可以根据自己的需求方便地执行分析。可以通过仅指定几个参数并执行主命令来调用一次全面的分析运行。或者,用户可以单独执行管道的步骤。初次使用的用户和专家都可以根据自己的需求方便地执行分析。可以通过仅指定几个参数并执行主命令来调用一次全面的分析运行。或者用户可以单独执行流程的步骤。
—————————————————-
我的使用体验:RnBeads的输入文件是主流比对软件(bismark等)比对后提取甲基化信息的结果文件或者甲基化芯片的结果文件,做的主要是数据过滤、可视化、基因组注释和差异分析,但出图质量不怎么样,不够美观。
有能力的还是根据RnBeads的结果文件或者不使用RnBeads自己用甲基化分析工具可视化。
#—————————————————-
安装RnBeads包
BiocManager法安装
install.packages("BiocManager")library(BiocManager)BiocManager::install("RnBeads")packageVersion("RnBeads")tools::dependsOnPkgs("RnBeads") # 查看依赖的包都有哪些
conda法安装
如果是在Linux系统下操作,建议直接使用conda安装RnBeads包,因为这个包的依赖确实非常多,conda的一个优点是能够一键式帮你解决依赖包/库的安装问题。
注意:现在R版本已经更新到4.0以上,RnBeads团队将包更新到的2.9.3版本,其依赖于4.0以上的R,但conda现在还不支持下载2.9.3版本的RnBeads,所以说conda有一定的滞后性。
# 新建一个R4.0的环境(optional)conda create -n r4.0# 安装4.0版本的Rconda install -c r r=4.0# 或者同步创建和安装R环境和Rconda create -n r4.0 r=4.0# 查找RnBeads现有的版本conda search -c r *rnbeads*# 安装2.8.0版本的RnBeadsconda install -c bioconda bioconductor-rnbeads=2.8.0
整个安装过程通常需要10至15分钟;根据需要安装或更新软件包的数量,以及计算机的速度和互联网连接,在极少数情况下,安装可能需要超过一个小时才能完成。安装脚本终止后,您可以使用以下命令验证安装是否成功。如果安装遇到报错请继续往下看,里面有本人安装时的踩坑记录。
library(RnBeads)rnb.run.dj()
安装基因组注释文件
如果您正在处理来自老鼠或大鼠基因组或最近的人类基因组组合的亚硫酸氢盐测序数据,您可能还需要安装bioconductor相应的软件包,如下所列,这取决于您想使用哪种基因组组合。目前仅支持 the human (hg19, hg38), mouse (mm9 and mm10) and rat (rn5) genomes。
# 下载RnBeads支持的基因组注释文件,最好提前查看下支持下载的基因组# 如果测序物种是人类,就下载hg19/hg38BiocManager::install("RnBeads.hg19")# 如果测序物种是小鼠,就下载mm9/mm10BiocManager::install("RnBeads.mm10")
参考:https://bioconductor.org/packages/3.13/bioc/html/RnBeads.html
安装依赖软件:Ghostscript
- Windows下
前往https://www.ghostscript.com/下载最新版的Ghostscript安装包
安装方法参考:https://rnbeads.org/data/installing_rnbeads.html,主要是要安装完后把安装位置添加到系统可识别的环境变量,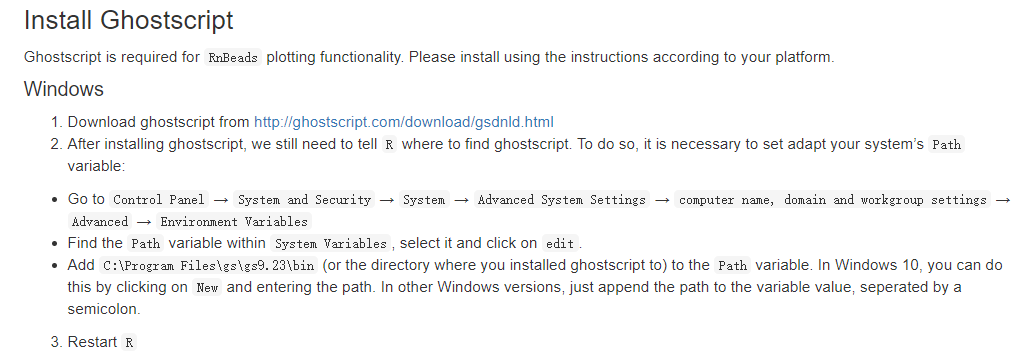
添加环境变量的具体方法如下
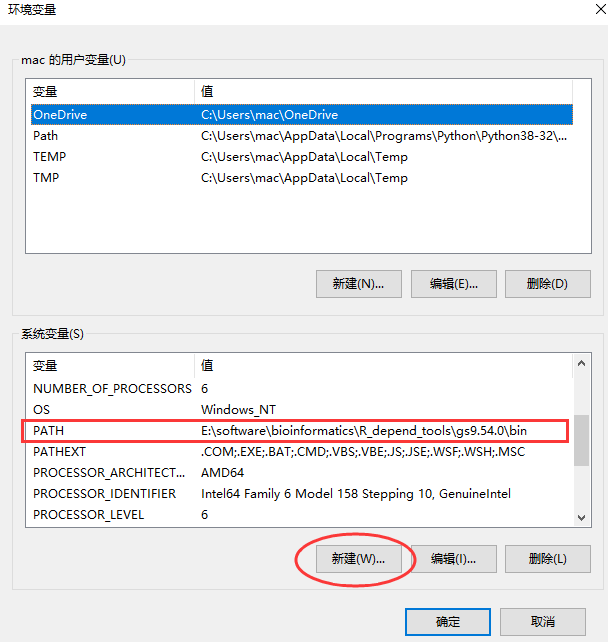
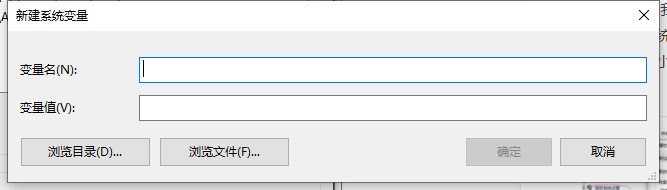
首先右键 “我的电脑/此电脑”图标,选择“属性”,选择“高级系统设置”,然后点击弹窗的“环境变量”,然后点击系统变量里的“新建(W)…”,然后填写变量值(Ghostscript 安装的位置)以及变量名。
这里有一个小细节,添加的环境变量名称必须是“PATH”,其他变量名貌似不行,反正我测试的一个名字就不行。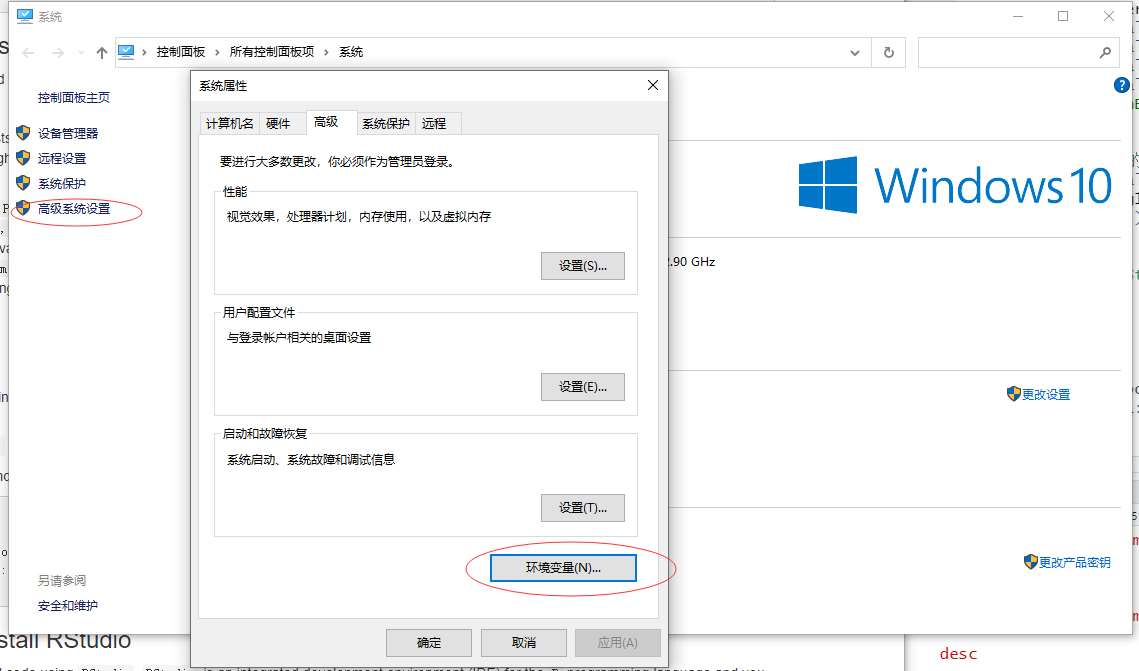
- Linux系统下
使用以下命令测试 Ghostscript 是否安装成功,如果安装成功了,运行完下面的命令后你的当前工作路径会产生一个plot图形文件# 安装依赖软件:ghostscriptconda install ghostscript
library(RnBeads)setwd(tempdir())rplot <- createReportPlot("test_gs")plot(1:20, pch = 1:20)off(rplot)
运行RnBeads
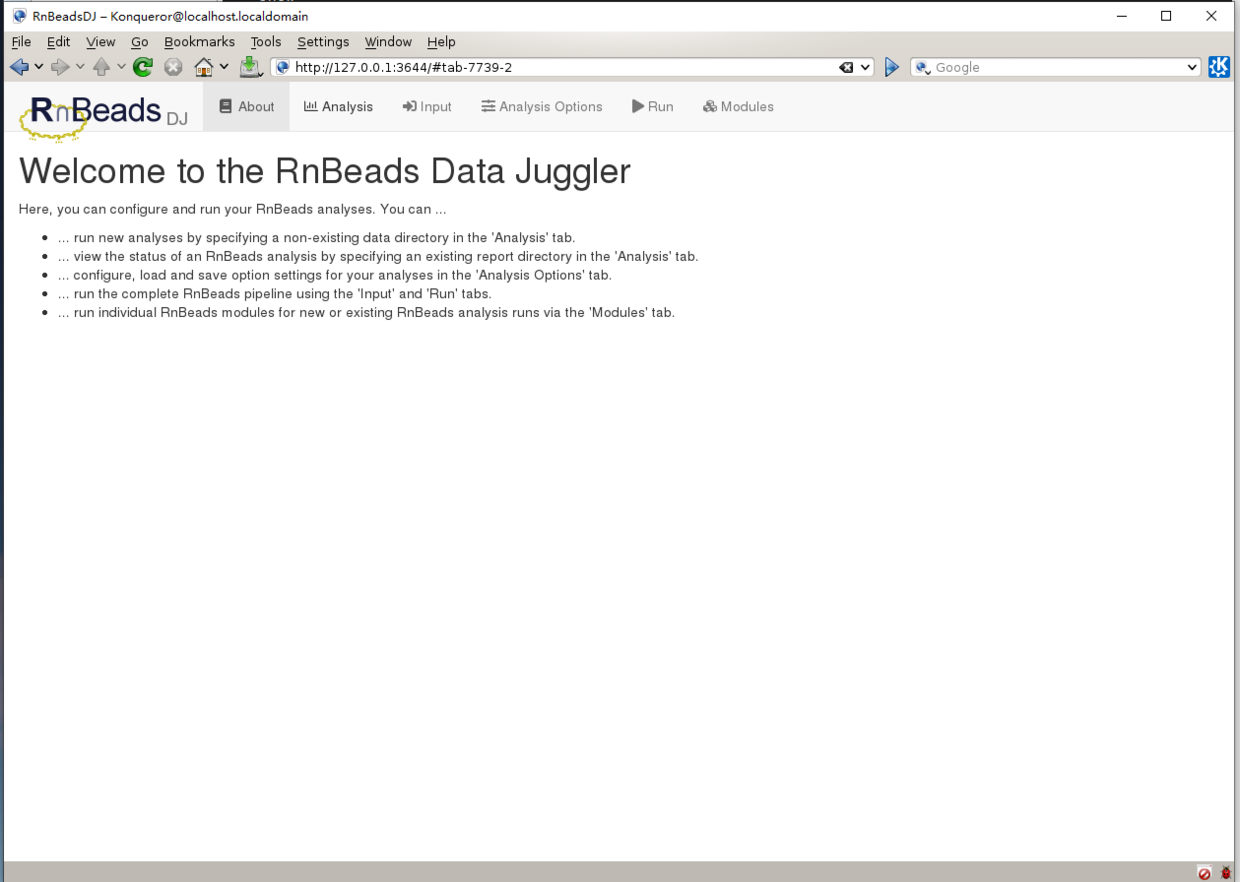
假设RnBeads及依赖项都已经安装好,使用“rnb.run.dj()”调出交互式的GUI(图形界面),接下来就是根据你的输入数据类型以及样本组别进行正确的点点点。
# suppressPackageStartupMessages(library(RnBeads))library(RnBeads)rnb.run.dj()
如何选择GUI图形页面的选项建议先看一看:
RnBeads的quickstart:https://medical-epigenomics.org/software/rnbeads/materials/data/tutorial/RnBeadsDJ/RnBeads_GUI_quickstart.pdf
现记录几个关键选项的设置:
- 数据输入
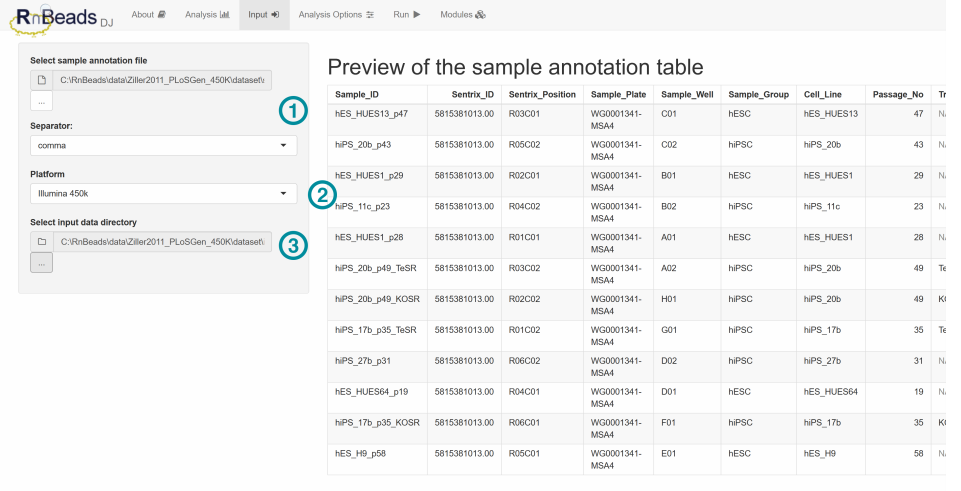
标记1:样本组别等信息;
标签2:数据由哪种测序平台获得的,可选项有甲基化芯片、BS-seq等;
标签3:输入文件的路径;
- Analysis options – loading option profiles
- Analysis options – general settings
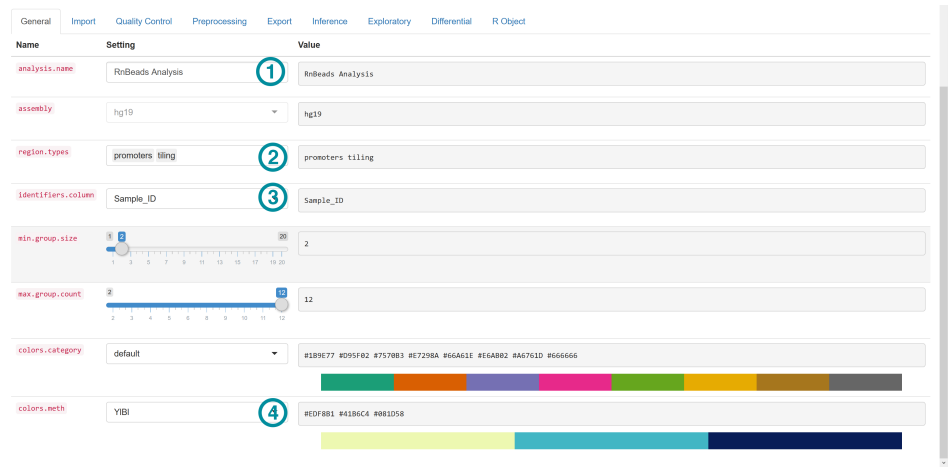
标签1: assign a reasonable name for the analysis.
标签2: select the region types for which methylation levels will be summarized in the analysis
标签3:select a column of the sample annotation table that will be used as sample identifiers,这是样本信息表里的样本唯一识别符,可以选择是样本名等没有重复值的一列的列名
标签4:select one of the predefined color schemes for categorical and methylation values
- Analysis options – differential methylation
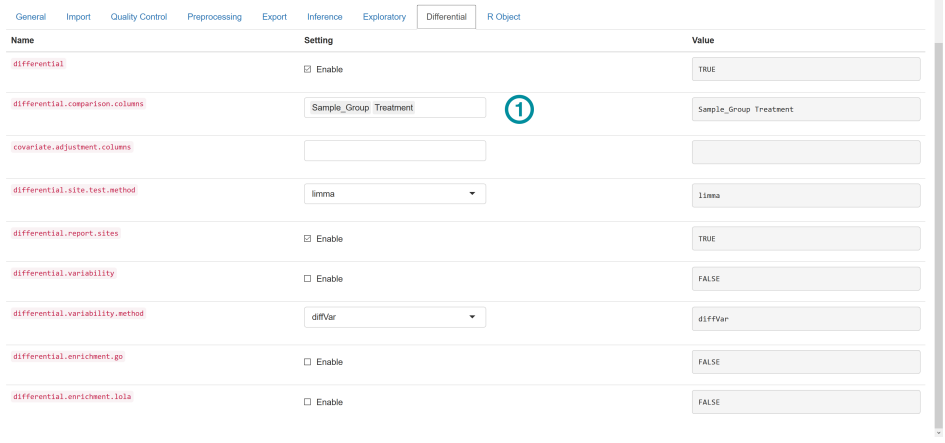
标签1:For our example, select “Sample Group” and “Treatment” as the columns that define sample groups that will be compared in the analysis. 即差异分析时设置的组别信息,比如你的样本处理信息以及样本来源信息这两列的列名。
如果全部参数正确,RnBeads运行完分析流程后会在R打印并且会自动打开网页版的分析报告,结果文件中的“index.html”是分析报告汇总。
2021-04-18 21:24:57 8.5 STATUS COMPLETED Saving RData2021-04-18 21:24:57 8.5 STATUS COMPLETED RnBeads Pipeline
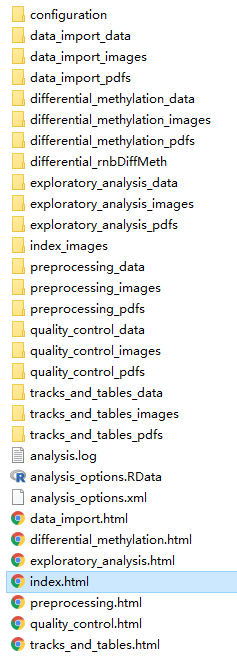

注:完整分析结果文件文件非常多且体积也很大,担心空间不足的可以研究提交任务时的参数,选择不输出拓展分析,差异分析时的一些结果文件。
安装时的报错记录
接下来记录一些安装时的报错合集,ps:这软件安装真的有点难
安装报错1:这个报错是由于rhdf5filters包在编译时出错,从报错上看起来是一个基于python的zstd包可能与当前环境的python版本或者R版本冲突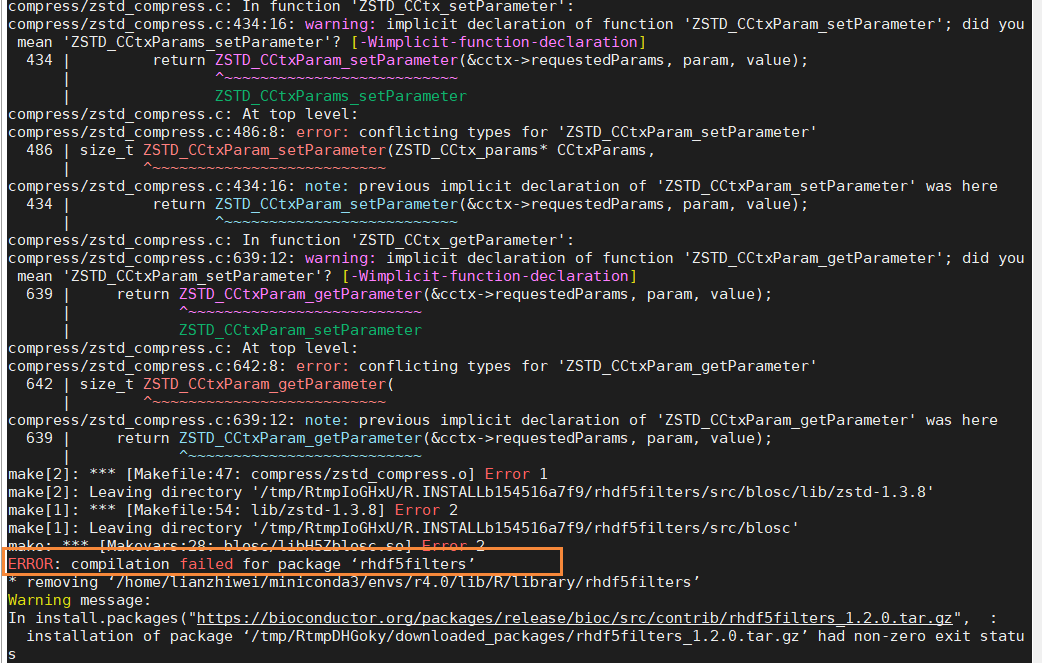
解决办法:
# 在R里面尝试了各种办法都是因为编译不成功安装不上,无奈尝试condaconda search *rhdf5filters*conda install bioconductor-rhdf5filters
tips:安装某些R包由于依赖包很多,或者更新不及时,导致安装过程会经常报错,只能说,见一个坑填一个,多点Google/bing,尝试使用conda安装R包
参考:https://blog.csdn.net/ARPOSPF/article/details/84997200
报错2:Ghost没有安装成功
运行到一半又说Ghost没有安装
Warning: Error in logger.error: Could not create file. GhostScript was not found
下面的报错原因也是Ghost没安装好,不过这是在Linux系统上发现的一个报错
/usr/bin/gs: /usr/local/MATLAB/MATLAB_Runtime/v901/bin/glnxa64/libtiff.so.5: no version information available (required by /lib64/libgs.so.9)
使用上面提到的办法重新安装试试
报错3:调用交互式页面出错了
> library(RnBeads)> rnb.run.dj()Listening on http://127.0.0.1:5927Error in utils::browseURL(appUrl) :'browser' must be a non-empty character string
貌似是RnBeads的版本问题,尝试下载源代码安装,因为conda和BiocManager还没支持最新版(2.9.3)
https://www.biostars.org/p/489273/
shell命令行从bioconductor下载安装压缩包
wget -c https://bioconductor.org/packages/3.13/bioc/src/contrib/RnBeads_2.9.3.tar.gz
R命令行下
install.packages("RnBeads_2.9.3.tar.gz", repos = NULL)# 安装完成后查看版本是否更新> packageVersion("RnBeads")[1] ‘2.9.3’
结果还是同样的报错,可能是我远程连接服务器的问题,防火墙等等原因。
报错4:有关rlang包的一个报错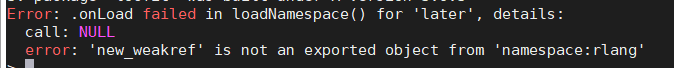
答案参考:https://github.com/r-lib/rlang/issues/900
报错5:shiny包引起的问题
> rnb.run.dj()Error in get(name, envir = asNamespace(pkg), inherits = FALSE) :object '%AND%' not found
见:https://www.biostars.org/p/492356/,实测有些人会出现有些人不会出现
GUI交互式页面提交任务后运行的可能报错记录:
- 如果运行时出现报错,可以先去rnbeads官网FAQ查找一下答案:https://rnbeads.org/faq.html
比如下面的这个报错就是因为提交参数时差异分析的组别设置不合理或错误。
如果交互式页面里的General那一页提示没有“gradient.rect”函数,证明 plotrix 包没有安装,为什么这里才发现,因为加载RnBeads包时也没有提示。
BiocManager::install("plotrix")
报错说行名重复了,证明交互式页面里选择的
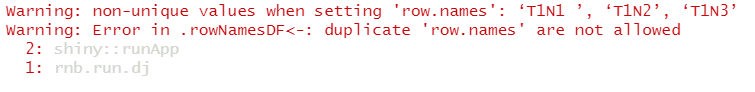
- 发现在服务器(Centos系统)下使用RnBeads,弹出来的交互式页面没法选中,就无论我点哪里都没有反应
。尝试用命令行远程打开服务器端的Firefox,输入RnBeads交互式页面的网址,如下图中圈起来的网址,居然可以打开交互式的GUI!
注:RnBeads也有命令行执行模式,即一条一条R command执行,但既然可以交互式(点点点),就先记录交互式的操作流程先,等有需要在记录命令行模式。
RnBeads用于BS-seq data分析的教程搬运
5 Analyzing Bisulfite Sequencing Data
RnBeads可以分析WGBS、RRBS类型的数据。用RnBeads分析亚硫酸氢盐测序数据的一般结构与Infinium数据相同。然而,问题出在细节上。以下是关键区别:
- 除了人类甲基化数据,RnBeads还通过其配套包(RnBeads.mm9,RnBeads.mm10,RnBeads.rn5)支持小鼠和大鼠甲基化组分析。
- 加载是通过bed文件输入执行的。
- 预处理过程中不进行归一化。
- 在质量控制和批效果报告中没有亚硫酸氢盐测序的控制探针。相反,覆盖率信息被用来评估实验质量。
- 当使用亚硫酸氢盐测序数据时,某些类型的分析将花费更长的时间,因此在标准分析中禁用。其中,对Infinium数据集的profiles模块中得到的不可靠样本和站点进行过滤的贪婪切算法和偏差图。
5.1 Data Loading
RnBeads希望测序数据采用bed格式或者具有类似的坐标文件格式。The data.source argument of the rnb.run.import() and rnb.run.analysis() functions for
analyzing bisulfite sequencing data requires a vector or list of length 1, 2 or 3 containing:
the directory where the methylation bed files are located;
the sample annotation sheet;
the index of the column of the sample annotation sheet that contains the names or full paths to the bed files;
In case only the first element of data.source is given, RnBeads expects the bed files and sample annotation to be located in the same directory. The sample annotation file should contain token ”sample” in the file name. The sample annotation should contain the names of the bed files relative to the specified directory. There should be exactly one bed file for each sample in the directory.
In case only the sample sheet is provided as the second element of the data.source list (the first element can be set to NULL), the provided sample sheet should contain absolute paths to the bed files. If the third element is missing, RnBeads attempts to find the file name-containing column automatically.
bed文件预计包含每个个体位点(CpG)的甲基化和定位信息,这些信息包括染色体、坐标(起始和结束坐标)和基因组链。甲基化信息应该包含任意两个数字,表明甲基化事件(#M -为特定位置测序的胞嘧啶数目)、未甲基化事件(#U -为胸腺嘧啶测序的数目)和总事件(#T = #U + #M)在特定位置被测量的数量。这可以用以下任何形式指定:
any two measurements among #M, #U and #T;
#T and the methylation percentage: #M/(#U+#M) × 100;
由于对于这类甲基化数据没有明确的标准,规范可能会很棘手。可能存在的不确定性的例子有:
Are the coordinates 0-based or 1-based?
What chromosome and strand notation is used?
Are the CpGs listed for both strands or just one?
Are the coordinates on the reverse strand shifted?
Is the methylation percentage specified in the interval [0,100] or [0,1]?
Many more questions may arise. 因此,不能保证RnBeads正确加载每种格式。可能装载亚硫酸氢盐测序数据最方便和安全的方法是使用为特定工具和管道量身定制的格式预置。这些预置在包选项’ import.bed.style ‘中指定。以下是目前可用的预设示例,(支持的软件输出):
’BisSNP’、’EPP’、’bismarkCov’、’bismarkCytosine’、’Encode’;
格式要求请参考给出的:https://bioconductor.org/packages/release/bioc/vignettes/RnBeads/inst/doc/RnBeads.pdf。
剩余内容待更新。

若有收获,就点个赞吧
0 人点赞
