File formats
https://cgmaptools.github.io/cgmaptools_documentation/file-formats.html
cgmaptools convert
想要使用cgmaptools进行DNA甲基化数据分析,首先得获取输入文件,如果你上一步进行序列比对时使用的软件是BSseeker,那么输入文件就是比对后进行甲基化信息提取获得的文件。
命令行输入cgmaptools convert --help,即可查看cgmaptools有哪些文件转换命令。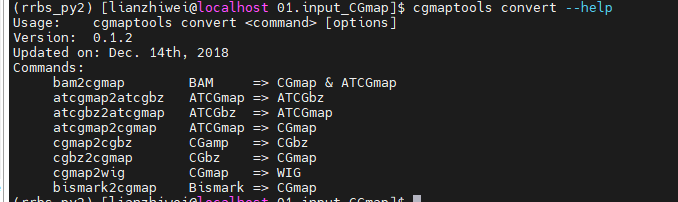
Description : File format coversion.
* Example :* WGBS-BAM to CGmapcgmaptools convert bam2cgmap -b WG.bam -g genome.fa --rmOverlap -o WG* RRBS-BAM to CGmapcgmaptools convert bam2cgmap -b RR.bam -g genome.fa --rmOverlap -o RR* ATCGmap to ATCGbzcgmaptools convert atcgmap2atcgbz -c WG.ATCGmap.gz -b WG.ATCGbz* ATCGvz to ATCGmapcgmaptools convert atcgbz2atcgmap -c WG2.ATCGmap.gz -b WG.ATCGbz* CGmap to CGbzcgmaptools convert cgmap2cgbz -c RR.CGmap.gz -b RR.CGbz* CGbz to CGmapcgmaptools convert cgbz2cgmap -c RR2.CGmap.gz -b RR.CGbz* CGmap to WIGcgmaptools convert cgmap2wig -i <CGmap> [-w <wig>] [-c <INT> -b <float>]* bismark output to CGmapcgmaptools convert bismark2cgmap -i bismark.dat -o output.CGmap
因为我习惯使用bismark这个软件进行比对和甲基化信息同期,所以会对以下两种进行解释
- bismark2cgmap:input is CpG report format
- bam2cgmap:input is BAM format
bismark2cgmap:CpG report format
**cgmaptools convert bismark2cgmap --help**
- input format:
- bismark比对软件提取甲基化信息的输出文件:Genome-wide cytosine methylation report(全基因组胞嘧啶甲基化报告),文件后缀为”*.CpG_report.txt”;
- 需要按照染色体坐标排序;
- 如果是以bismark生成的甲基化信息文件” .bismark_bt2_pe.C*_report.txt”为输入,因为bismark是不管有没有reads覆盖到这个位点,默认会把基因组上的所有的C位点的甲基化状态保存在这个文件中,那分析前需不需要过滤掉那些没有reads覆盖到的位点?
示例格式:
- output format:CGmap Format
- Example
每列含义:chr1 G 3000851 CHH CC 0.1 1 10chr1 C 3001624 CHG CA 0.0 0 9chr1 C 3001631 CG CG 1.0 5 5chr1 G 3001632 CG CG 0.9 9 10

重点解释:
第六列为甲基化水平,取值为[0,1]或者NA;
第七列为支持位点为甲基化的reads;
第八列为这个位点被多少条reads覆盖(检测),俗称覆盖度
Tools:shell语言
脚本名称:cgmaptools_convert_bismark2cgmap.sh
目的:批量使用**cgmaptools convert bismark2cgmap**命令进行文件格式转换
Usage:bash cgmaptools_convert_bismark2cgmap.sh <input file path>
#!/bin/bash# 目的:cgmaptools convert bismark2cgmap# 输入文件类型:bismark提取甲基化位点获得的Genome-wide cytosine methylation report格式文件,文件后缀为"*.CpG_report.txt";# 输入文件路径input_bismark_CpGreport=${1}# 输出文件路径output_CGmap=${input_bismark_CpGreport}/bismark2cgmap# Determine if the folder exists. If it does not, create a new one,shell语言要求中括号内的每个空格都是必须的if [ ! -d ${output_CGmap} ];thenmkdir -p ${output_CGmap}elseecho "output direction is already exist."fi# 获取输入文件列表并读取ls ${input_bismark_CpGreport} |grep "CpG_report" >bismark_CpG_calling.Filelistcat bismark_CpG_calling.Filelist | while read filename;doif [ "${filename##*.}" = "gz" ];then# 如果输入文件为"gz"压缩格式prefix1=$(echo ${filename} | awk -F '.gz' '{print $1}')# -c, --stdout write on standard output, keep original files unchangedgunzip ${input_bismark_CpGreport}/${filename} -c >${input_bismark_CpGreport}/${prefix1}.CpG_report.txtcgmaptools convert bismark2cgmap -i ${input_bismark_CpGreport}/${prefix1}.CpG_report.txt -o ${output_CGmap}/${prefix1}.CGmaprm ${input_bismark_CpGreport}/${prefix1}.CpG_report.txtelse# 如果输入文件为非压缩格式prefix2=$(echo ${filename} | awk -F '.txt' '{print $1}')cgmaptools convert bismark2cgmap -i ${input_bismark_CpGreport}/${filename} -o ${output_CGmap}/${prefix2}.CGmapfidonerm bismark_CpG_calling.Filelist
实测转换后的文件行数不变。
bam2cgmap:BAM format
运行cgmaptools convert bam2cgmap -h查看命令参数
Usage:cgmaptools convert bam2cgmap -b <BAM> -g <genome.fa> -o <prefix>
Notice:
For BS-Seeker2, CGmapTools seamlessly comparable with its BAM.
For Bismark, CGmapTools comparable with single end mode;
and comparable with v0.8.2 and older version of bismarks for paired-end.
Bismark changed FLAG strategy since v0.8.3, which raise unexpected count of nucleotides.
* For BSmap, we can not guarantee generating right CGmap from our experience.
输入的bam文件要求:
- 输入的bam文件必须按照染色体坐标排序
使用samtools对bam文件排序
samtools sort [-l level] [-m maxMem] [-o out.bam] [-O format] [-n] [-T tmpprefix] [-@ threads] [in.sam|in.bam|in.cram]参数:-l INT 设置输出文件压缩等级。0-9,0是不压缩,9是压缩等级最高。不设置此参数时,使用默认压缩等级;-m INT 设置每个线程运行时的内存大小,可以使用K,M和G表示内存大小。-n 设置按照read名称进行排序;-o FILE 设置最终排序后的输出文件名;-O FORMAT 设置最终输出的文件格式,可以是bam,sam或者cram,默认为bam;-T PREFIX 设置临时文件的前缀;-@ INT 设置排序和压缩是的线程数量,默认是单线程。example:samtools sort -o output.bam -O bam -@ 8 input.bam
输出文件:三种压缩格式的文件,分别是
- prefix.ATCGmap.gz
- prefix.CGmap.gz
- prefix.wig.gz
```bash
prefix.ATCGmap.gz
1 G 3735063 CHH CT 0 0 0 1 0 0 0 0 1 0 1.00 1 A 3735064 — — 1 0 0 0 0 1 0 0 0 0 na 1 C 3735065 CHG CT 0 1 0 0 0 0 1 0 0 0 0.00
prefix.CGmap.gz
1 C 3009136 CHG CC 0.00 0 2 1 C 3009137 CG CG 1.00 2 2 1 G 3009138 CG CG 1.00 2 2 1 C 3009139 CHG CA 0.00 0 2 1 G 3009141 CHG CT 1.00 2 2
prefix.wig.gz
variableStep chrom=1 3009136 0.00 3009137 1.00 3009138 1.00
**实战**<br />**输入:bismark比对软件比对的输出文件:bam格式**<br />_更新:cgmaptools have not support the lastest bismark align output to convert to CGmap, but the author say it still useful if you remove the "--rmOverlap" flag。_文件转换代码:vim cgmaptools_convert_bam2cgmap.sh<br />目的:批量转换```bash#!/bin/bash# 参考基因组路径RefGenome=/bismark/hg19/hg19.fafor sorted_bam in $(ls | grep ".bam$");doecho "cgmaptools converting ${sorted_bam}"sampleID=$(echo ${sorted_bam} | awk -F '_merge' '{print $1}')cgmaptools convert bam2cgmap -b ${sorted_bam} -g ${RefGenome} -o ${sampleID}done
Tips:
- 假如输入的BAM文件是来自
bismark,那么运行cgmaptools convert bam2cgmap理论上会生成三种类型文件,但是如果添加--rmOverlap参数后就只能生成一种类型文件,这可能是因为cgmaptools开发的时间比较早,而bismark早期版本的输出的BAM文件和目前已更新好多代的BAM文件不太一样,所以就会报错。所以作者也建议,运行时不添加--rmOverlap参数。
如果没有这个参数,则获得的CGmap格式下包含CG/CHG/CHH三种胞嘧啶状态,其中经统计,CG数目会多于使用bismark进行methylation calling出来的CG数目,通过查看获得的CGmap文件格式,我的猜测是--rmOverlap参数导致正负链的位点同时被统计了,但是没有确切的依据,GitHub上也找不到答案。
番外:统计一下有多少个CpG位点
# CGmap文件格式1 C 3009136 CHG CC 0.00 0 21 C 3009137 CG CG 1.00 2 21 G 3009138 CG CG 1.00 2 21 C 3009139 CHG CA 0.00 0 21 G 3009141 CHG CT 1.00 2 21 C 3009143 CHH CT 0.00 0 2zcat T1N1_Overlap.CGmap.gz | awk '{if($5=="CG")print}' | wc -lzcat T1N1_Overlap.CGmap.gz | awk '{if($5=="CG" && $8>=2)print}' | wc -l

若有收获,就点个赞吧
0 人点赞
