说到三维基因组学,大家可能会问: 什么是三维基因组学?三维基因组学是一门研究基因结构与功能关系的组学。那我们就从基因结构与功能相适应的角度来看看C技术是如何一步一步发展成现在这个样子的。下图是我们分子生物学一个经典的启动子增强子模型,普遍认为enhancer会募集很多转录因子以及转录辅助因子结合到启动子区域形成一个环状结构(loop)来调控基因的表达。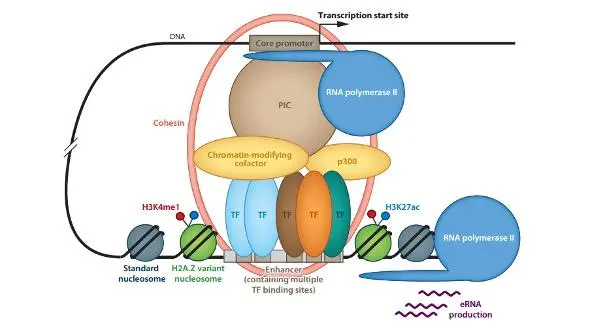
那么我们先回归一个最最基本的问题,那就是什么是基因?在测序之前,我们对基因的概念是模糊的,基因并不是简单的ATCG的组合,基因上游有启动子,启动子上游有调控元件,如果说基因不需要调控,那么我们把基因提取出来,放到试管里自然就会产生蛋白,这显然是不可能的,那么就产生了第二个问题,基因表达到底是通过什么样的方式被激活的?
我们知道DNA是双螺旋结构,双螺旋的DNA缠绕在八聚体的核小体上,经过进一步折叠缠绕形成了30nm的微小管结构,而微小管经过进一步折叠,最终形成了染色质结构。如果说我们人类细胞核内的DNA拉直,大概有2m的长度,但是最终折叠到几十微米的细胞核中,如此致密的结构会不会拉近基因之间在空间的距离,增大基因间的接触从而调控基因的表达?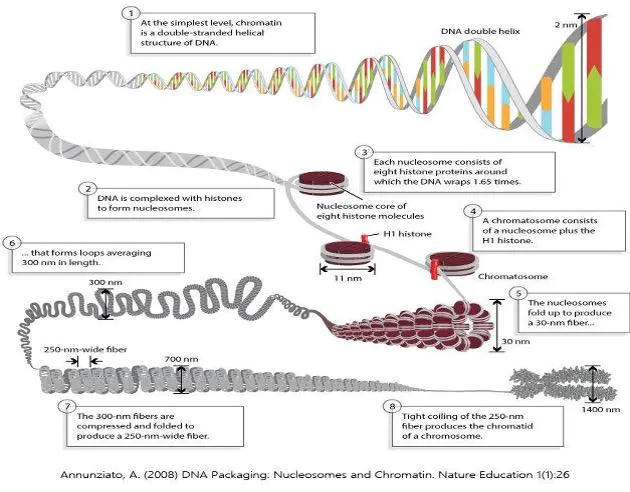
在1993年,人类基因组刚提出来,人们对基因的理解也不是很深入,但大家普遍关心的一个问题就是基因是怎么被调控的。当时有两种假说,一种是线性模型,也就是基因组线性位置较近的基因元件之间更容易互相调控。另一种是环状模型,也就是说线性距离较远的基因组元件通过成环,使其在空间更为接近,从而影响基因表达。Cullen实验室对于第二种假说更为感兴趣,那么如何证明这一假说呢?
Cullen实验室用病毒质粒包裹一个enhancer 和其调控的基因如下图,A是enhancer ,C是A调控的基因,AC之间夹着基因B,在ABC之间均有限制性内切酶酶切位点。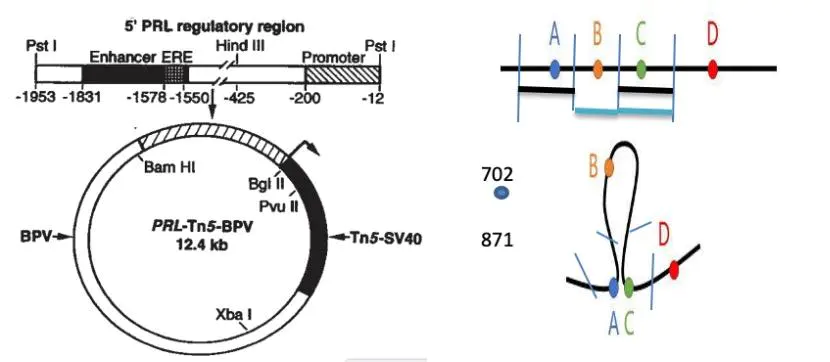
如果基因表达调控是线性的话,那么经过我们的限制性内切酶酶切,平末端补平,跑胶之后,B,C所在的酶切片段会更容易链接,BC链接的酶切片段大概为871bp,因此跑胶后,会在871bp处形成明亮的光带。相反,如果基因表达调控是环状结构的话,AC 所在的酶切片段更容易链接,跑胶后,会在702bp处形成明亮的光带,而最终通过跑胶的结果证实了基因的表达调控呈现一种环状的结构。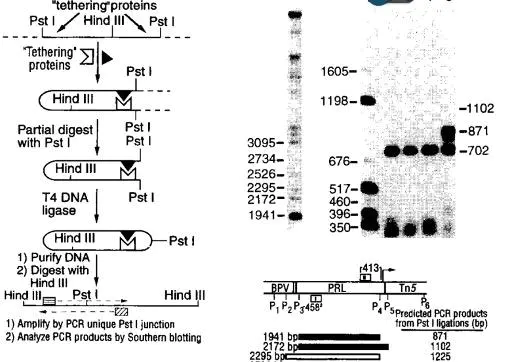
那么我们再次回顾一下cullen实验室的做法:
1. 构建病毒载体,包裹基因与其调控元件;
2. 利用HindIII限制性内切酶进行酶切;
3. 组蛋白消化;
4. 利用T4 DNA 连接酶进行连接;
5. 通过PCR扩增;
6. 跑凝胶电泳;
时隔8年,2002年dekker实验室在cullen实验室的基础上提出了3C的概念 ,从而打开了三维基因组的大门。那么3C 技术又是如何做的呢?它在真核细胞中增加了甲醛交联的步骤。这样,就是将细胞固定住,使其能够维持生前最后一刻的形态,通过限制酶切割和平末端补平来获得基因组上互作的片段,通过蛋白酶消化,以便后续进行测序。通过设计一对引物,来钓取我们研究的感兴趣的互作位点,最后通过凝胶电泳来验证这两点是否互作。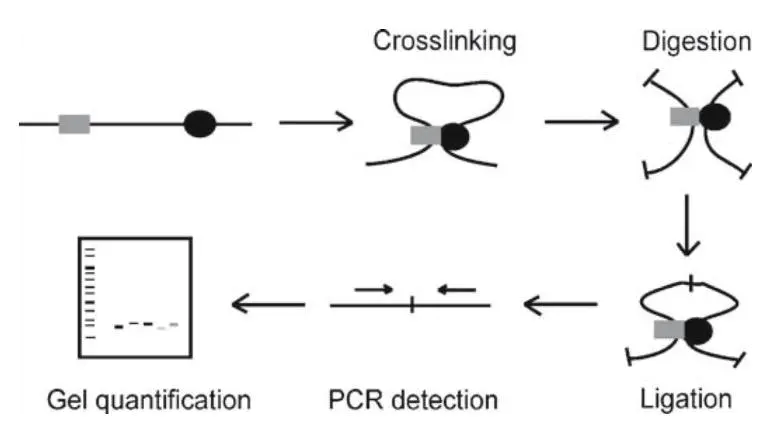
4C技术 是在3C的基础上增加了双酶切位点,促使其成环,这样在后期只需要设计一个引物就能获得一个位点与多个位点的互作关系。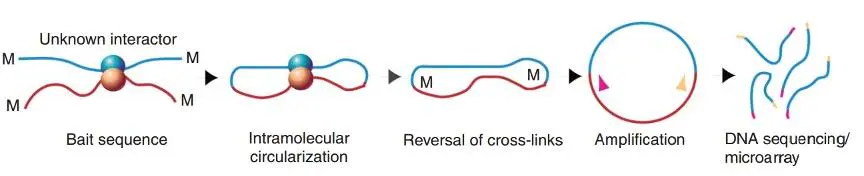
而5C技术是基于3C技术,对于感兴趣的一堆基因,设计多对引物,通过PCR扩增,这样的话,就能知道多对多的互作,特定基因互作网络的验证。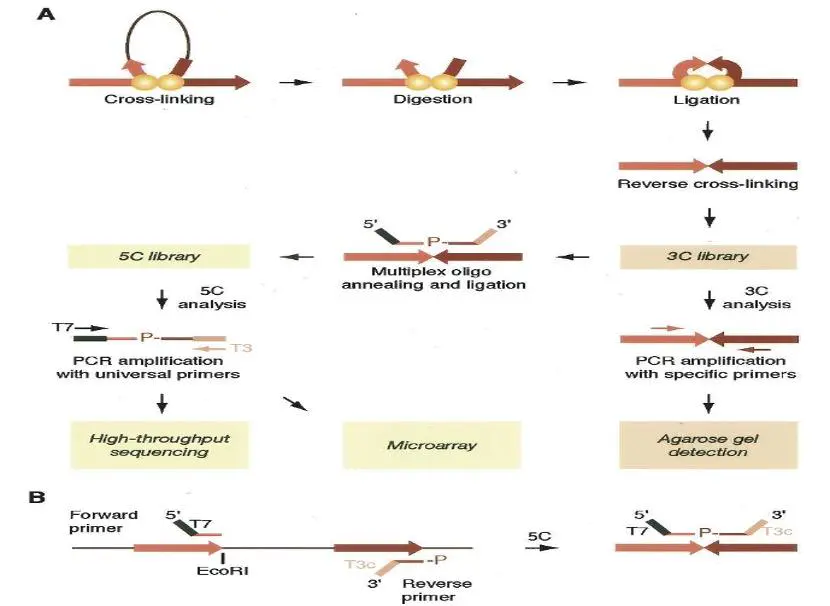
由于设计引物人力物力消耗过大,到了2009年,dekker实验室果断的放弃了引物的设计,最大化的发挥了酶切位点的作用,通过生物素来钓取互作的片段。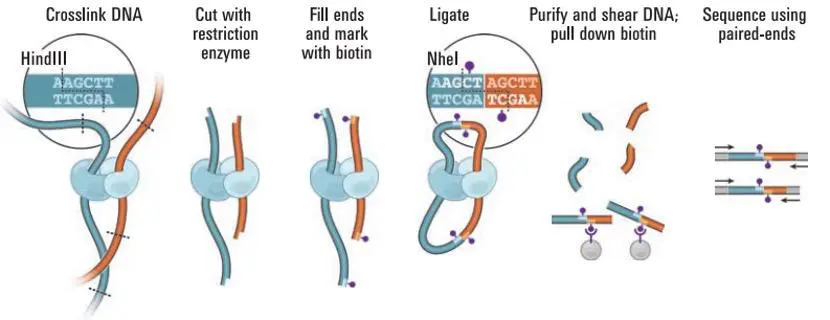
那么Hi-C 技术是怎么做的呢?
首先,利用甲醛交联,固定细胞的形态,通过限制性内切酶进行酶切,末端补平后,加生物素,进行平末端连接,组蛋白进行消化后通过磁珠进行钓取,最终获得的两端线性距离较远的互作片段。对这些互作片段进行测序,过滤,比对,我们最终得到了Hi-C 的互作矩阵,每一个点,代表着基因组两个位置的交互强度,而从这些互作矩阵中,通过特定的算法,我们能够获得基因组的高级空间结构。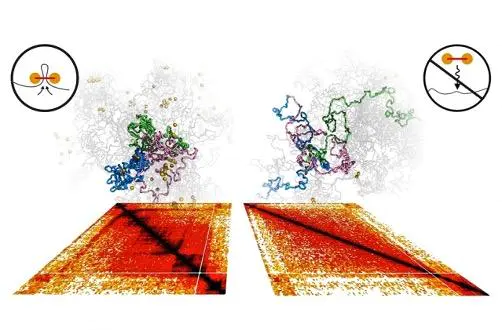
Capture Hi-C 在Hi-C 文库的基础上,对感兴趣的区域像外显子流程那样,用杂交的方式,利用特定的探针去杂交,这样就能特征性的富集感兴趣区域的互作关系。Capture Hi-C的优点是用更少的数据量获取更为精确的互作信息。那测序的对象来说,对于同样的数据量,Hi-C 测的是全基因组,而capture Hi-C 测的却是感兴趣的少量基因,我们把测序的数量比作粥,测序的基因比作僧人,那么相对capture Hi-C 而言,Hi-C面临的问题就是僧多粥少了。
对C技术有了一个大概的印象之后,那么大家可能会想,这些C技术各有什么优缺点?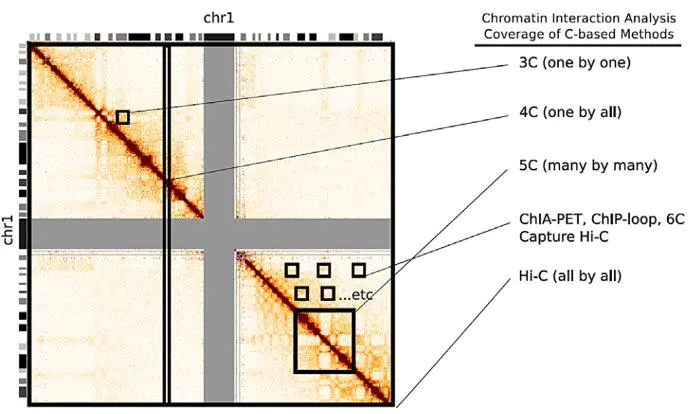
3C技术,得到的是一对一的互作关系,如果我推测出一个基因与肌原细胞相关的基因myog互作,那么测全基因组肯定是不划算的,可以拿3C技术进行验证 ,这个基因是否与myog互作。如果我只关心一个癌症易感突变位点,想知道它都与那些位点发生了互作,那么我就可通过4C技术来获取这个位点的所有互作信息。Capture Hi-C 技术基于所想即所得的前提,可以捕获我们关注的基因区域。Capture Hi-C 是我们心中已有沟壑的情况下的一个精准打击,而Hi-C则相当于在信息不明的情况下的一个地毯式搜查。
在介绍Hi-C的历史时,我们提到Hi-C 数据的分析时基于矩阵的,提到矩阵,就要提到划bin。那么什么是划bin,划bin就是基因组按照一定的大小划分成n等份,那么我们将获得nXn的互作矩阵。举个例子来说,如果我们将基因组划分成2份,分别为A0,A1,那么A00代表的是A0内部的互作信号。A01 代表的是A0与A1的互作情况。我们最终会得到一个2X2的矩阵。如果我们将基因组划分成4份,我们得到的将会是一个4X4的交互矩阵。划分成8份,得到的是8X8的交互矩阵(如下图)。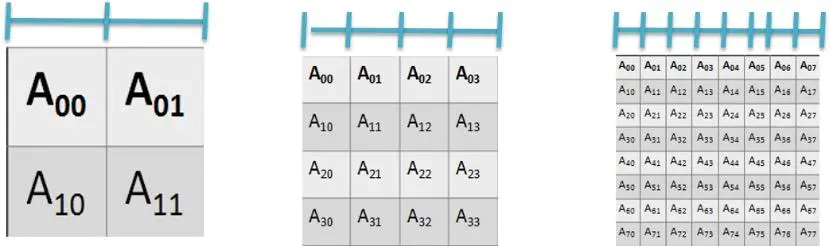
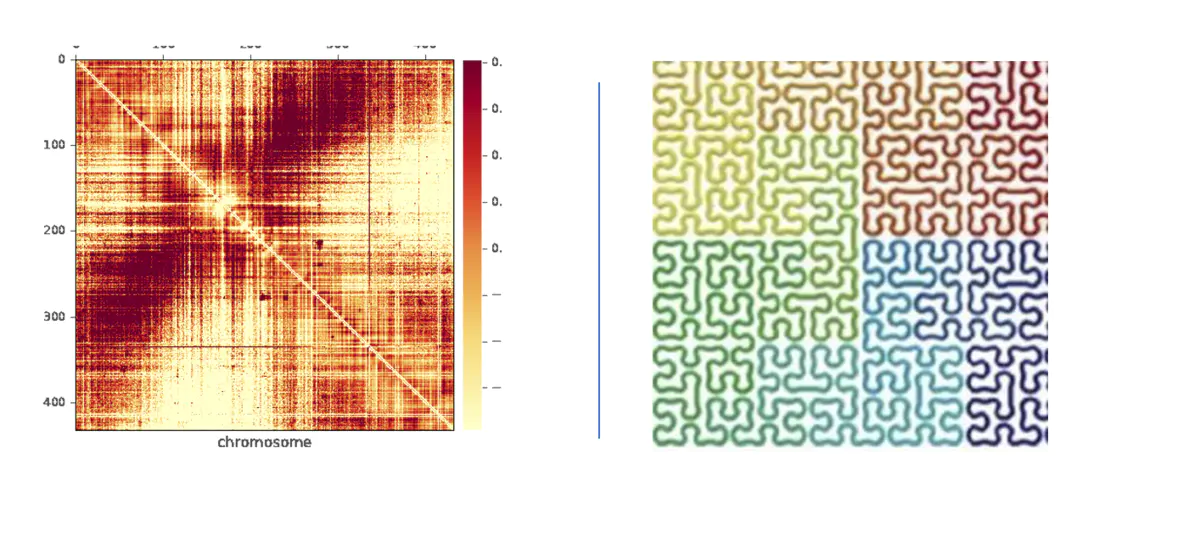
划bin越小就越能接近细微的结构。但是我们不能将bin无限划小,因为通过以往文献的经验来讲,在数据量没有达到一定的水平时,为了研究更为细微的三维基因组结构,我们贸然将bin划小, 这就类似于,在像素没有达到一定的水平,我们贸然的放大图像最终也只会得到一个模糊的影像,并不能获得精确的信息。
达到不同的数据量要求,我们可以分析不同的三维基因组结构,而不同数据量衡量的指标叫做分辨率,我们Hi-C的分辨率也是借鉴了光学分辨率的概念,也就是达到了某一分辨率,我们能够观测到某一结构。
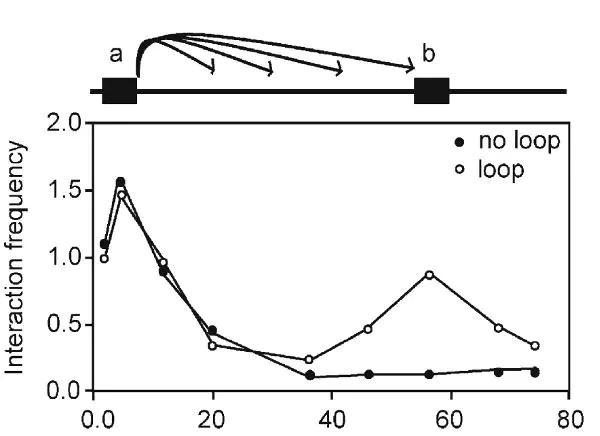
那么对于Hi-C 而言,都有哪些不同层级的结构呢?在80年代后期,通过荧光原位杂交发现在细胞核中,每条染色体都占据着独立的区域,有着明显的边缘。Hi-C 可以通过互作的矩阵模拟染色体的三维空间结构。而对每一条染色体进行分析,我们发现每条染色体可以分为两个部分一个是活跃的区域(A compartment)一个是不活跃的区域(B compartment)。再进一步对compartment进行研究,我们发现每个compartment内部都有一些小的拓扑结构域(TAD),在这些拓扑结构域内部,基因间的互作比较频繁,而拓扑结构域之间的互作则比较少。深入到这些拓扑结构域的内部,我们发现在这些拓扑结构域内部总有一些基因区域之间的交互信号非常强烈,这些称之为loop。
简单介绍了一下Hi-C的各个高级结构,我们来看看我们具体是怎样识别这些结构的呢?
这是一个Hi-C 染色体的三维建模,通过我们的Hi-C 矩阵,我们可以获得每个bin的交互特征,Pastis通过计算每个bin之间的欧式距离,来定位每个bin的空间坐标,这样我们获得了每个点的坐标(x,y,z),之后通过pymol这位好搭档来进行可视化。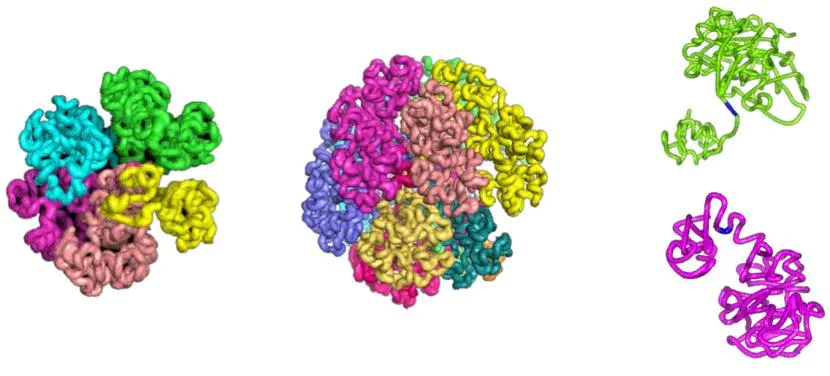
通过对原始矩阵的观察,我们发现原始矩阵中在热图上呈现的是格子状,反映在数据上是这么一个情况,那么我们怎么形象话这种现象呢?让我们切换到电影帮派斗争场景,每个帮派在各个区域都有眼线,如果我们把热图中每个bin比作一个人的话,那么每个点都是每两个人联系的次数,我们发现总有一堆人与不同区域的另外一堆人联系的比较密切,那么推理的话,这些人极有可能属于同一帮派。那么就有一个问题:1.是如何将这种现象放大,让我们看的更清晰一些,对于数据科学家来说拿起的武器就是先进行一个OE标准化之后做一个协方差处理(右图)。根据结果来看,这种方式挺好,我们的互作热图有着明显的红蓝格子分布。那么第二个问题就是如何将整个基因组根据其交互形式分成两类呢,这便运用到了我们的降维工具PCA(主成分分析),通过主成分分析,我们将基因组分成了两类。
红的代表一类,蓝的代表一类,那么这两类有什么特点呢?结合基因表达数据以及ChIP-seq数据,我们发现其中一类基因表达程度平均较高,富集了很多组蛋白结合位点,这一类是相当活跃的,科学家称之为A compartment,相反另一类不活跃,称之为B compartment。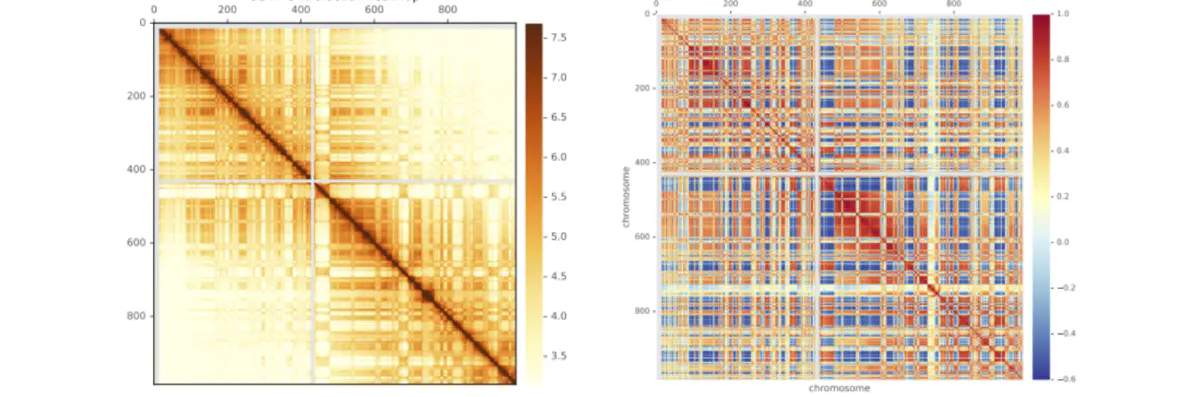
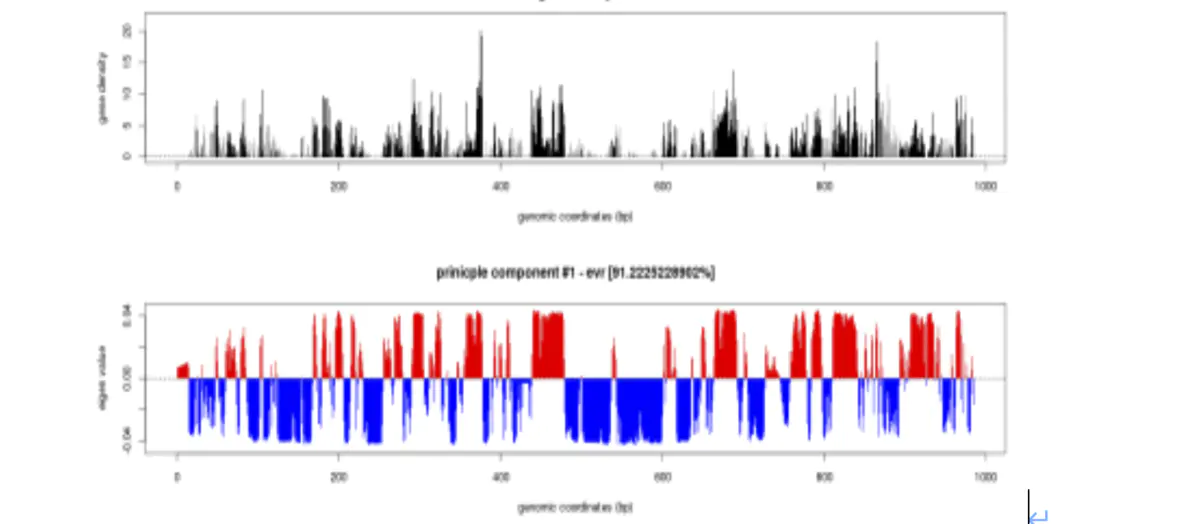
看完AB compartment 后,我们再来看看TAD的鉴定,TAD是因为科学家将整个热图沿着对角线平铺,发现有很多三角形的区域,这些三角形的区域在结构上,明显是这一团内的基因交互的比较明显嘛,而团与团还有明显的边界!这怎么理解呢,打个比方来说,就相当于我们安诺优达分为信息分析部门,项目管理部门,实验部门,部门之内的交流发生是很频繁的,而部门间的交流相对少一点。针对这一特点,dekker实验室识别TAD原理是在HiC互作矩阵中划定一个(500kb-3500kb)左右的范围,计算该范围内每个bin的互作数,作为未标准化的insulation score, 再计算bin的互作均值, 通过计算公式log2(insulation score/mean)得到标准化的insulation score。
为了确定TAD的边界,需要再次计算边界强度,对每一个bin设定一个更小的范围(100kb),计算每个bin的上游100kb所有bin的均值mean left 和下游每个bin的均值mean right,边界强度=mean left – mean right,当boundary strength 高于设定的delta阈值时,该reference Point就被认为是TAD 边界。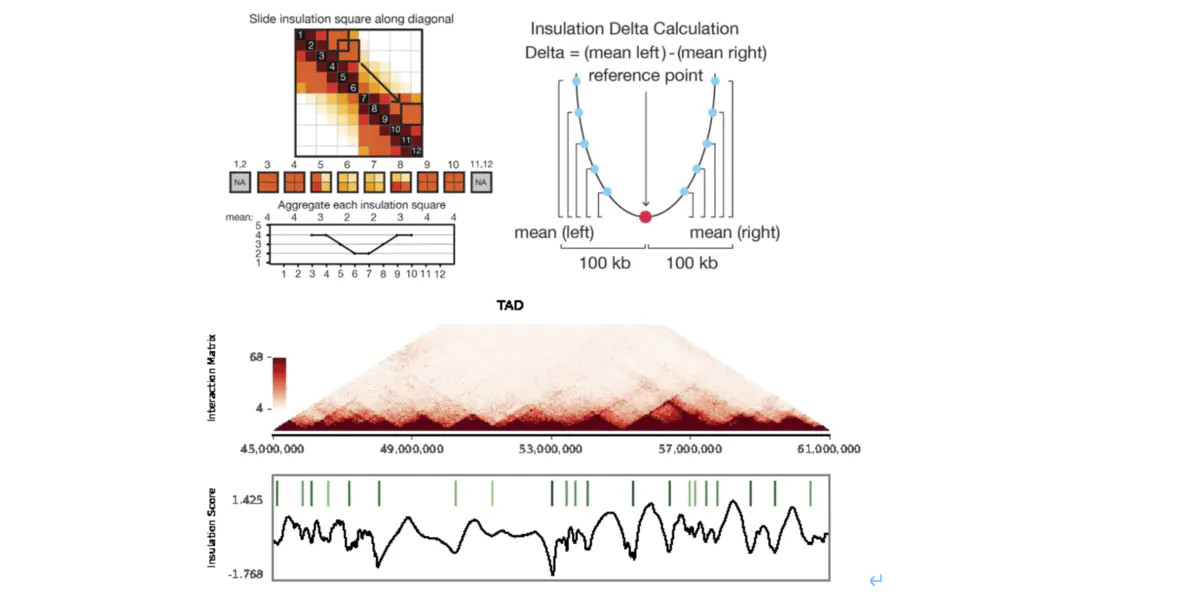
Loop是距离较远但是交互非常强烈的热点。对于loop的识别目前流行的有两种方法,一种是fit-Hic,另外一种是Hiccups。
我们先来看看hiccups,hiccups是运行在GPU上的软件,它是一种图形识别的算法。它会识别loop的热图特征。Hiccups识别的loops灵敏度低,准确性高。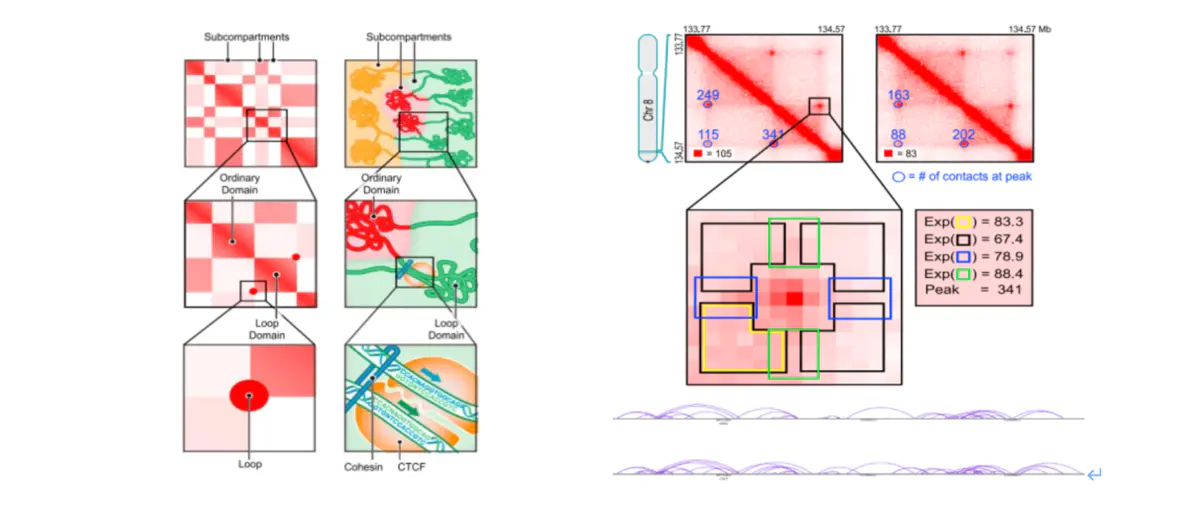
而fit-Hic 的识别方法是通过单调递减拟合来识别loop,fit-Hic 识别的loop灵敏性高,准确率低。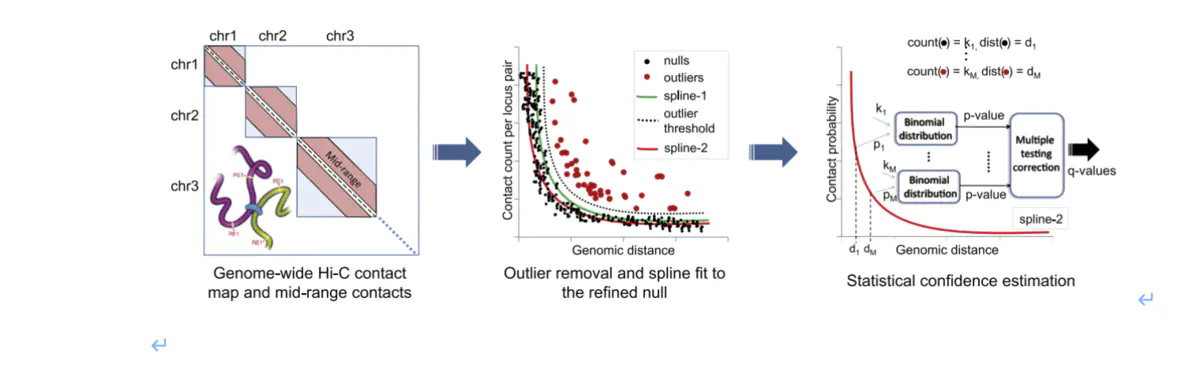
对于Hi-C而言,最重要的是要找结构上的差异。
首先,我们会考虑交互矩阵的差异: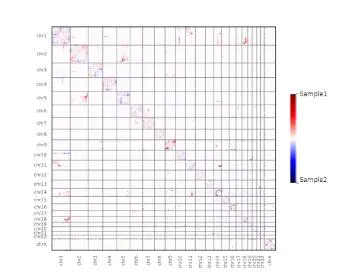
其次就是各个层级的差异比较:
首先是AB compartment的差异分析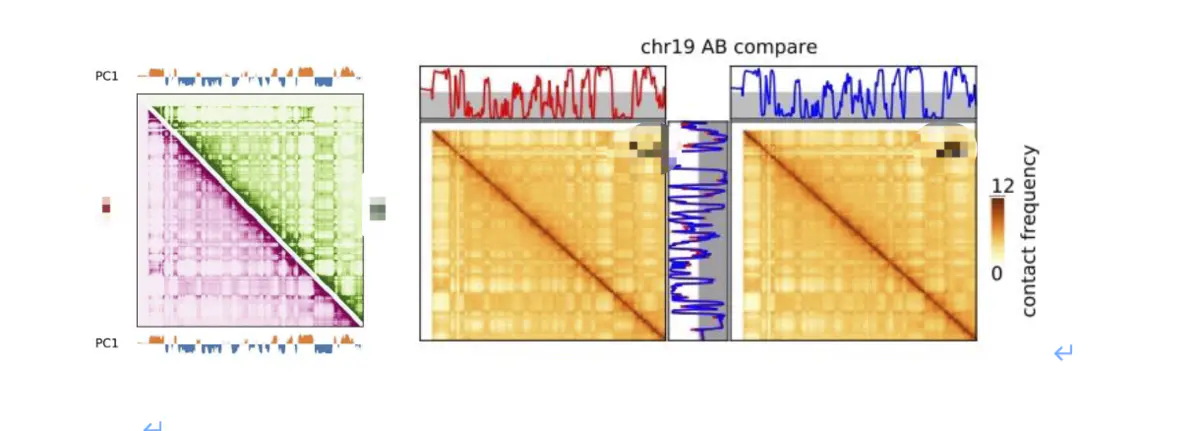
之后是TAD的差异分析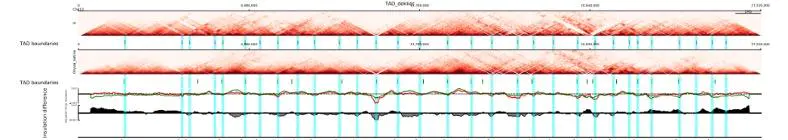
核心是loop层级的比较: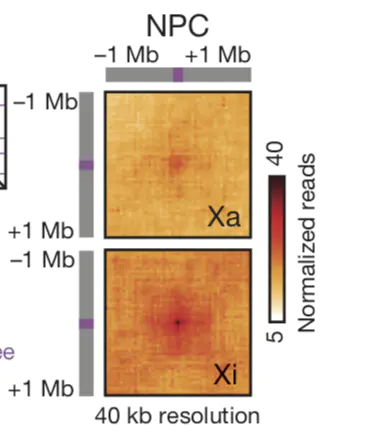
当然上面的比较只是最为常规的套路分析,而真正分析还是要结合课题的设计。
作者:XuningFan
链接:https://www.jianshu.com/p/a6a424cae892
来源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。

若有收获,就点个赞吧
0 人点赞
