学习目标:
- 探讨ChIP-seq数据质量低的来源
- 理解链交叉相关性( strand cross-correlation)
- 使用phantompeakqualtools计算交叉相关性和其他相关的质控度量值
-
ChIP-Seq质量评估
在下游分析前,最好是先对peak calling 后的ChIP-Seq数据进行质量评估。
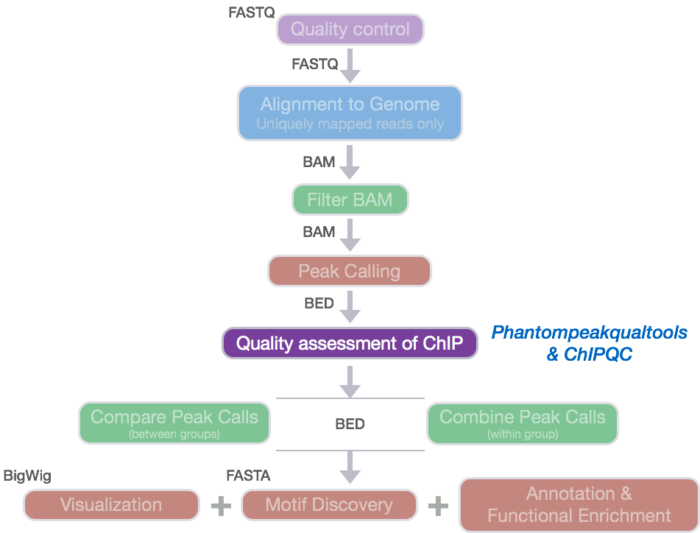
链交叉相关(Strand cross-correlation)
链交叉相关是一个有效的评估ChIP-Seq质量的方法,它不依赖于peak calling,而是基于ChIP-Seq实验。如果ChIP-Seq实验成功,DNA富集序列标签(蛋白质相互作用的序列)会在reads的双峰富集中产生显著的聚集。
产生reads的双峰富集的原因如下:
在ChIP-Seq实验中,DNA被片段化,蛋白质结合的片段会被免疫沉淀,所以产生了有蛋白质结合的DNA片段(fragments )。
DNA的正链从5’端开始被测序(如下图红色reads),DNA负链也从5’末端被测序产生如下图所示的蓝色reads。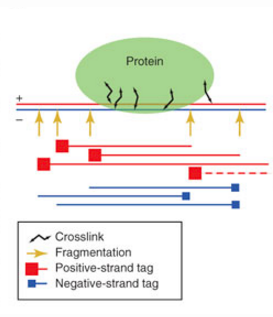
由于从DNA片段的5′末端测序,使 链reads的富集(下图中的蓝色部分)与负链reads的富集(下图红色部分)有少量的相互抵消区域。我们需要确定峰位移多少碱基数目可以在两个峰间产生最大的相关性。我们可以用交叉相关的度量值(cross-correlation metric)计算产生最大相关的位移。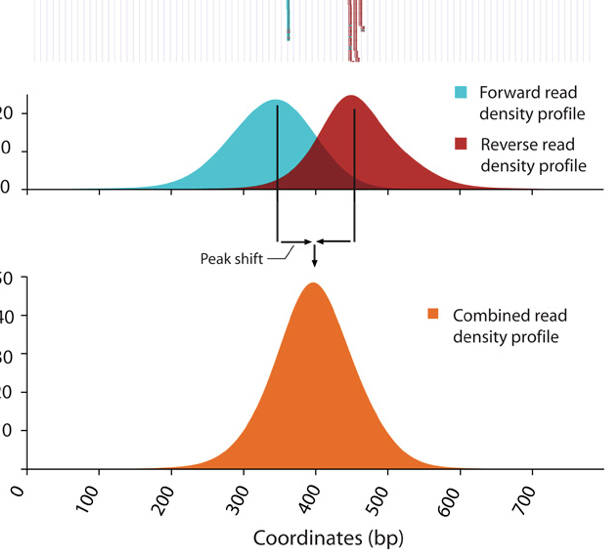
交叉相关性度量值
交叉相关度量是在Watson移动k个碱基后,计算Crick链与Watson链之间的Pearson线性相关。如下面的示意图:
首先在链位移为0时,两个向量之间的Pearson相关值为0.539。
在链位移5bp,两个向量之间的Pearson相关值为0.931。
继续移动这些向量,对于每个链位移计算一个相关值。
最后,我们将有一个每个碱基对移位与皮尔森相关值的对应表。这是针对每个染色体的每一个峰计算的,然后该值乘以一个缩放因子,再对所有染色体的值相加,就可以绘交叉相关值(y轴)相对于移位值(x轴)生成的交叉相关图。
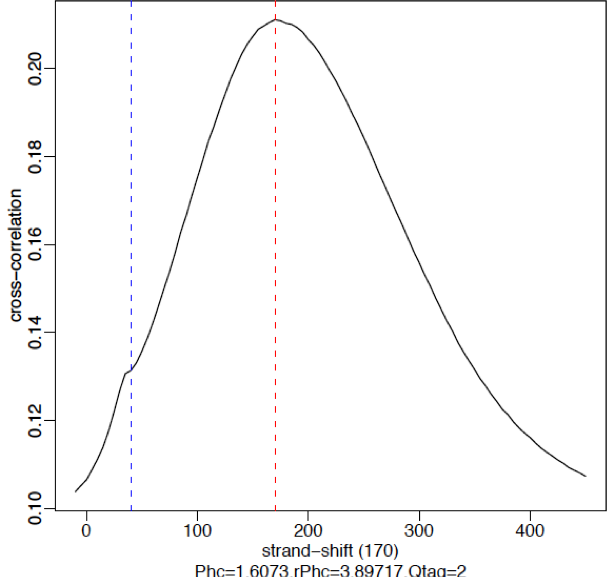
典型的交叉相关图会产生两个峰:一个富集峰与主要的片段长度(predominant fragment length)相关(高相关性),另一个与read 长度(read length)相关,这个峰也被称为虚幻峰(“phantom” peak)。 质量好的ChIP-Seq数据集倾向产生一个大的片段长度峰(fragment-length peak),下图展示了一个来自于人细胞CTCF(zinc-finger transcription factor)的强信号。如果有好的抗体,转录因子通常产生45,000~60,000个peaks。下图红色的垂直线表示主峰的真实位移,蓝色的垂直线处有一个小的起伏表示read lenngth。
- 下图是weaker signal的一个示例。这里Pol2的抗体不是很有效,有分散的峰。交叉相关图中有两个峰,一个是真实的峰位移(185-200bp),另外一个在read length。信号弱的数据集中read length的峰会成为主峰。
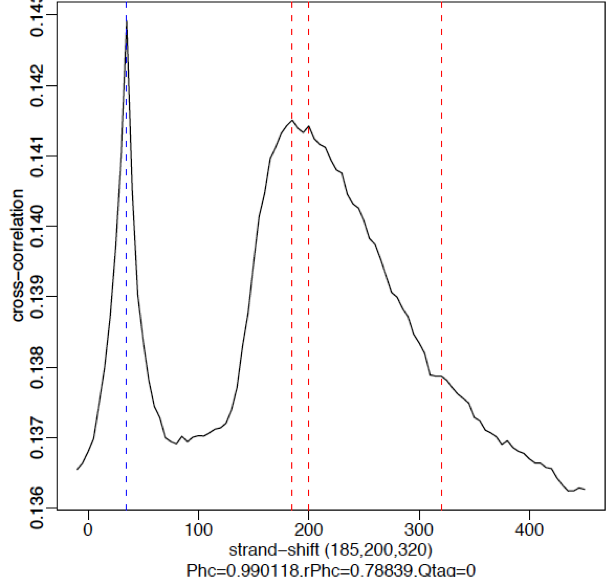
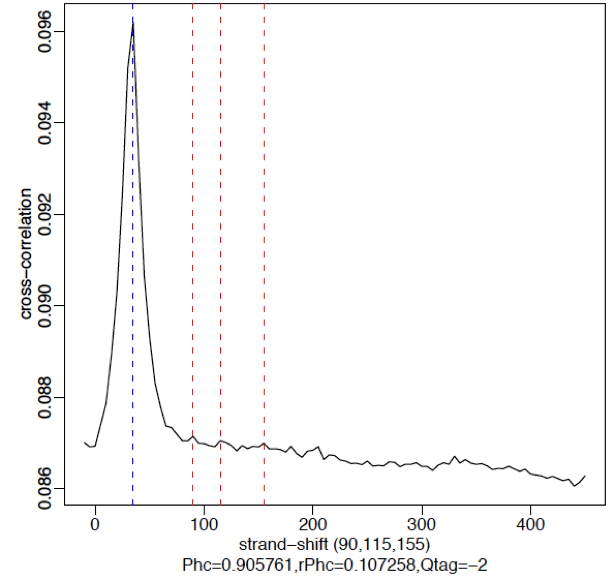
- 一个失败的实验产生的交叉相关图类似于input,在fragment length处很少或没有峰,在read length处有信号非常强的。这种现象的原因可能是在结合位点附近fragments没有显著聚集。
交叉相关性质量评估度量值
交叉相关谱图可以计算评估ChIP_Seq实验信噪比的度量值,并且实验设计确保fragment length准确。低信噪比和不准确的fragment length 表明ChIP-Seq实验可能有问题。
Normalized strand cross-correlation coefficent (NSC):
NSC是最大交叉相关值除以背景交叉相关的比率(所有可能的链转移的最小交叉相关值)。NSC值越大表明富集效果越好,NSC值低于1.1
表明较弱的富集,小于1表示无富集。NSC值稍微低于1.05,有较低的信噪比或很少的峰,这肯能是生物学真实现象,比如有的因子在特定组织类型中只有很少的结合位点;也可能确实是数据质量差。
Relative strand cross-correlation coefficient (RSC):
RSC是片段长度相关值减去背景相关值除以phantom-peak相关值减去背景相关值。RSC的最小值可能是0,表示无信号;富集好的实验RSC值大于1;低于1表示质量低。phantompeakqualtools
phantompeakqualtools 是一个用于计算ChIP-Seq数据富集和质量度量值的一个工具包。我们将使用该包来计算基于链交叉相关峰的主要插入大小(fragment length)和基于相对phantom peak的数据质量度量值。phantompeakqualtools是一个R包,依赖samtools。
下载phantompeakqualtools ```bash wget https://storage.googleapis.com/google-code-archive-downloads/v2/code.google.com/phantompeakqualtools/ccQualityControl.v.1.1.tar.gz tar -xzf ccQualityControl.v.1.1.tar.gz
cd phantompeakqualtools
查看README
less README.txt
**Linux下安装**```bashRinstall.packages("caTools", lib="~/R/library")
运行phantompeakqualtools
mkdir -p logs qualfor bam in bam_dir/sample1.final.bam bam_dir/sample2.final.bamdobam2=`basename $bam .final.bam`Rscript run_spp_nodups.R -c=$bam -savp -out=qual/${bam2}.qual > logs/${bam2}.Routdone
参数含义:
- -c: 比对过滤后的bam文件的全路径和名字
- -savp:保存交叉相关图
- -out:会产生数据集重要特征值的输出文件
输出文件解读
输出文件会产生一个tab分割的名为qual的文件,包含的信息如下:
- COL1:Filename:比对过滤的bam文件名
- COL2:numReads :有效的测序深度
- COL3:estFragLen:逗号分隔的交叉相关峰以相关性递减顺序排列的值
- COL4: corr_estFragLen: 逗号分隔的以递减顺序排列交叉相关值
- COL5: phantomPeak: Read length/phantom peak链位移
- COL6: corr_phantomPeak: phantom peak相关值
- COL7: argmin_corr:交叉相关最小的链位移
- COL8: min_corr:交叉相关最小值
- COL9: Normalized strand cross-correlation coefficient (NSC) = COL4 / COL8
- COL10: Relative strand cross-correlation coefficient (RSC) = (COL4 - COL8) / (COL6 - COL8)
- COL11: QualityTag: Quality tag based on thresholded RSC (codes: -2:veryLow,-1:Low,0:Medium,1:High,2:veryHigh)
我们最关注的值是第9列和第11列。
Cross-correlation plots
课程中的示例数据Nanog_rep1的交叉相关图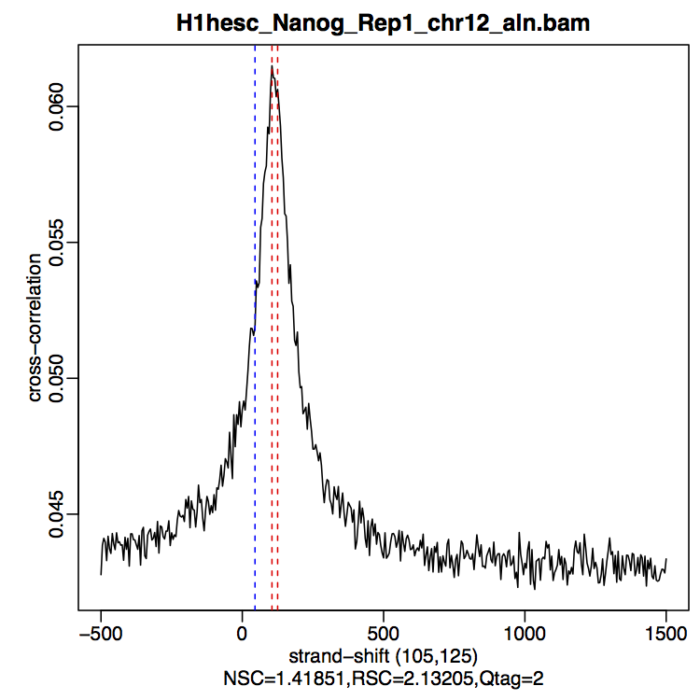
- 版权声明 本文源自 生信拾光, XP 整理
- 转载请务必保留本文链接:https://www.plob.org/article/24658.html

若有收获,就点个赞吧
0 人点赞
