说到三维基因组可视化,就不得不说一下washU browser(http://epigenomegateway.wustl.edu/)了。washU相当于IGV的升级版,一向被三维基因组CNS级别的文章所钟情,能够产生极其fancy的效果,是三维基因的可视化神器。
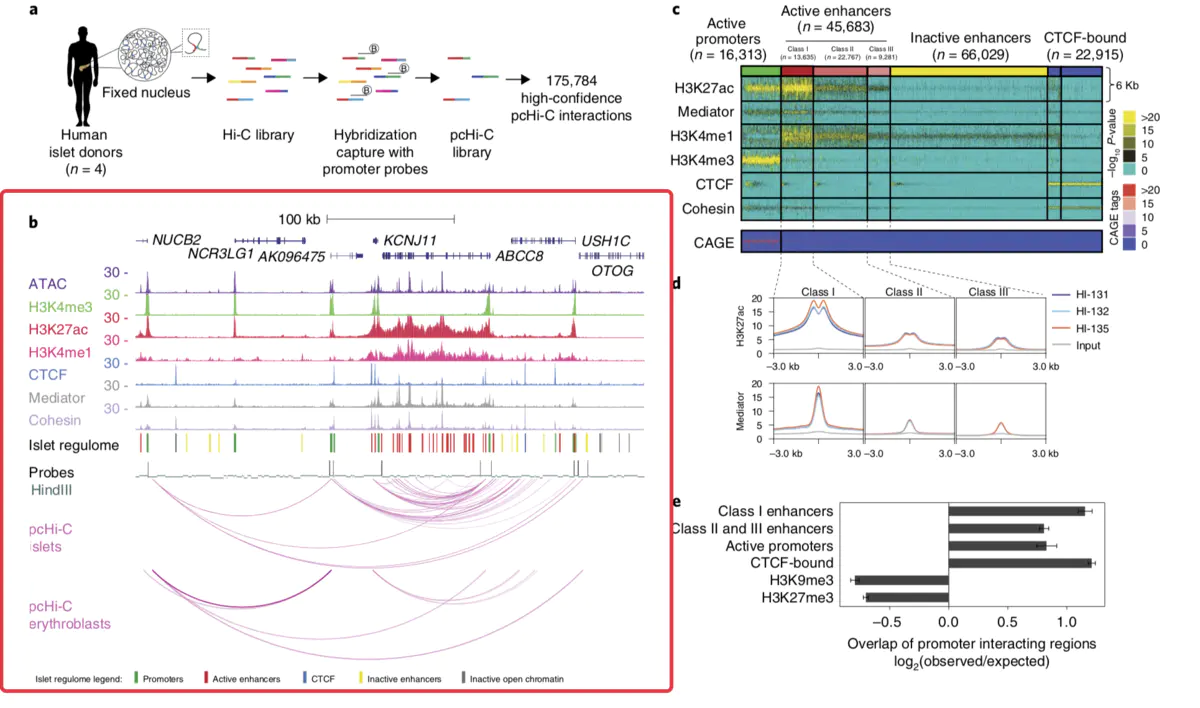
washU 目前支持包括人,小鼠,黑猩猩,斑马鱼等一系列物种。如下图所示。
你可以选择你需要的基因和版本进行可视化分析。
在选取了基因组版本号之后,你可以在搜索🔍框中,选择你所关注的基因或者SNP(如下图):
输入关注的基因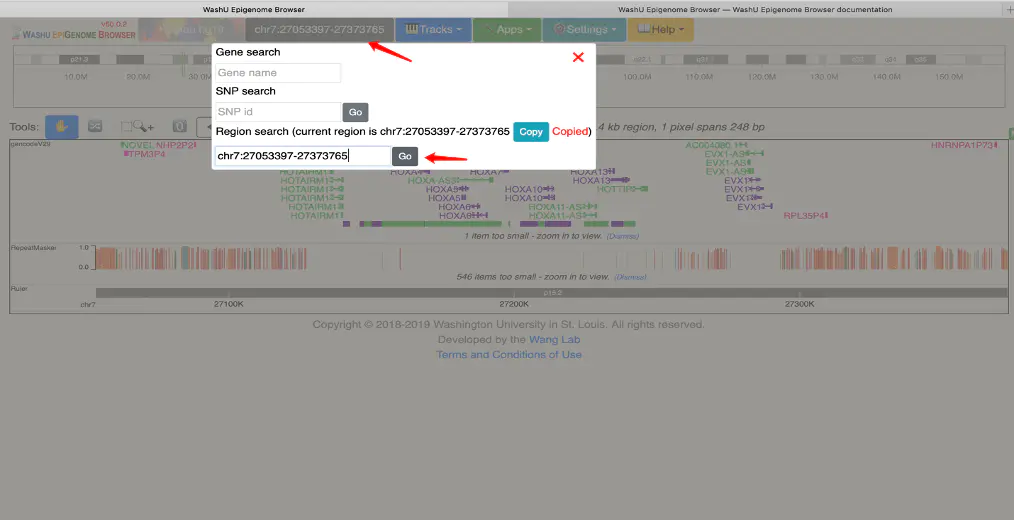
输入关注的snp
在选择完了关注的位点之后,你就可以通过view Local Tracks 导入你本地的文件。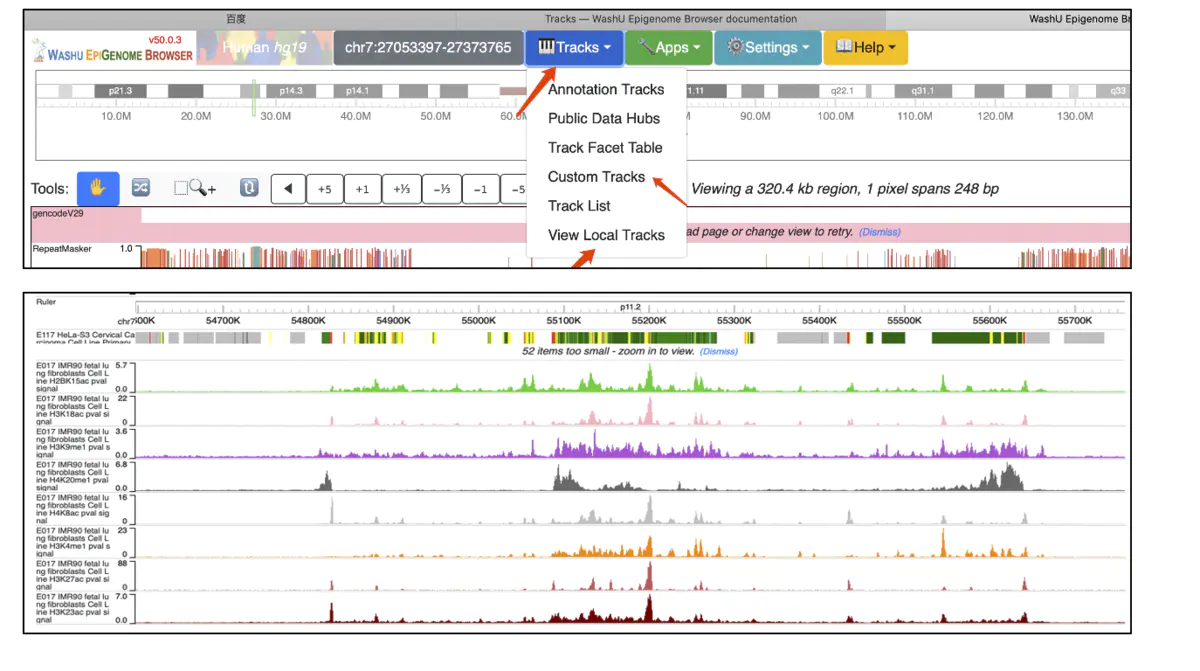
washU 支持的文件包括bigwig,bed,bam等等(如下所示)。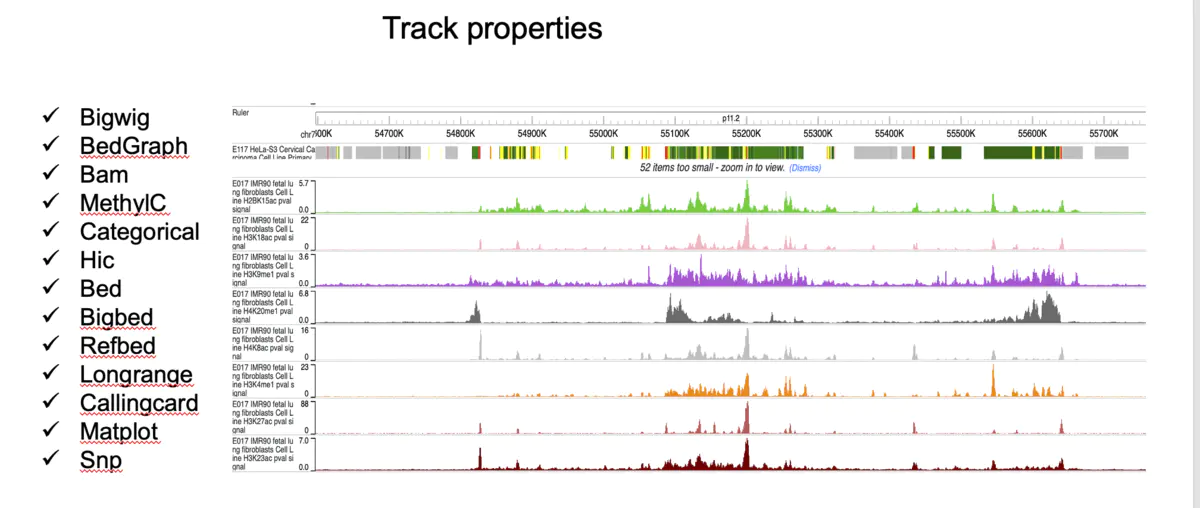
像bam,bigwig,bed文件可以导入washU,以峰图的形式呈现。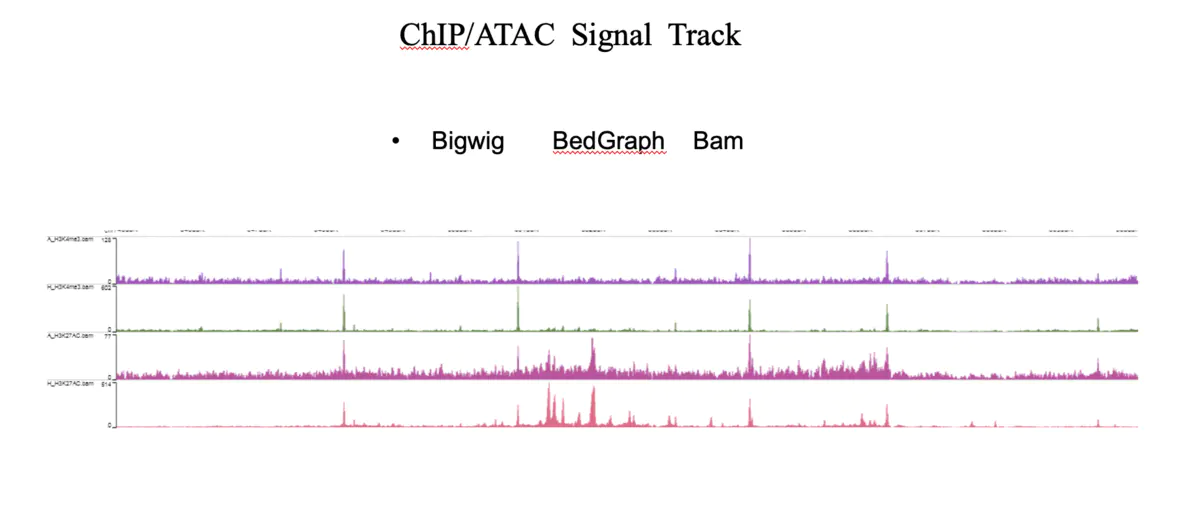
那么如何导入呢?有两种方式,一方面是刚刚所说的 本地上传 ,另一方面是通过URL提供数据,如下图: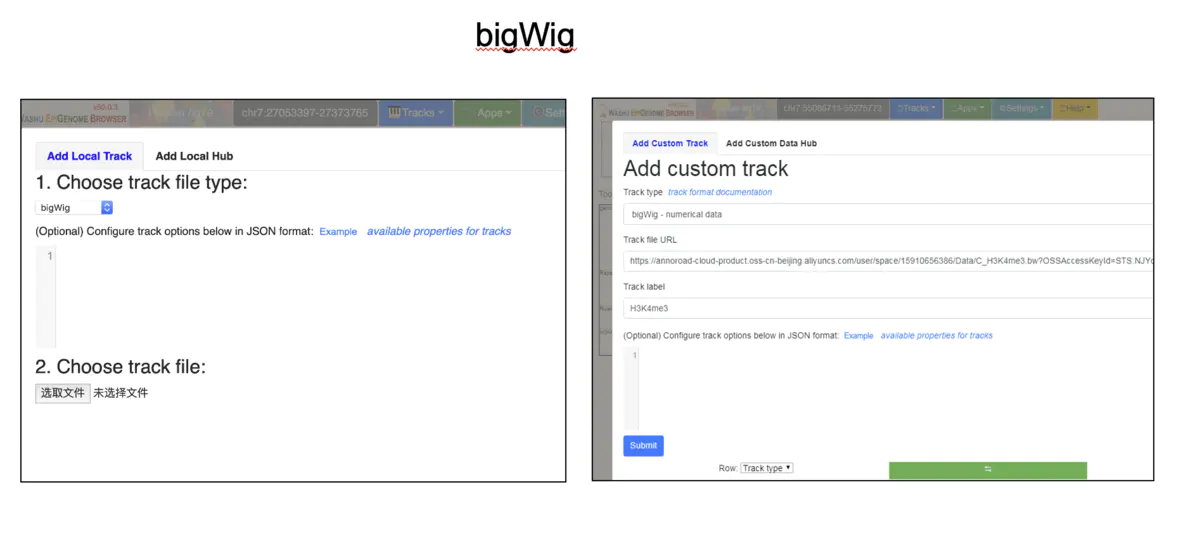
知道了数据类型和数据的上传方式,那么我们如何准备数据呢?
首先看bed文件(如下图):
包括4列: chr ,start,end ,value(如下图)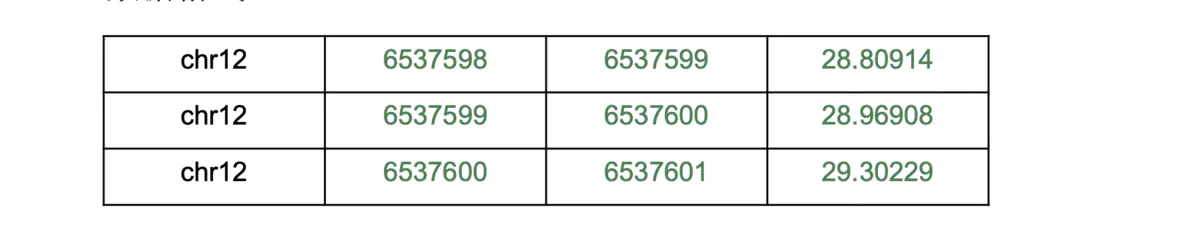
处理命令如下:
sort -k1,1 -k2,2n track.bedgraph > track.bedgraph.sorted #对bedgraph进行排序bgzip track.bedgraph.sorted # 对bedgraph进行压缩tabix -p bed track.bedgraph.sorted.gz #对压缩后的bedgraph 建立索
bgzip安装如下:
wget https://github.com/samtools/bcftools/releases/download/1.2/bcftools-1.2.tar.bz2
tar xvf bcftools-1.2.tar.bz2
cd bcftools-1.2
makemake install
samtools sort test.bam > test.sorted.bam
samtools index test.sorted.bam
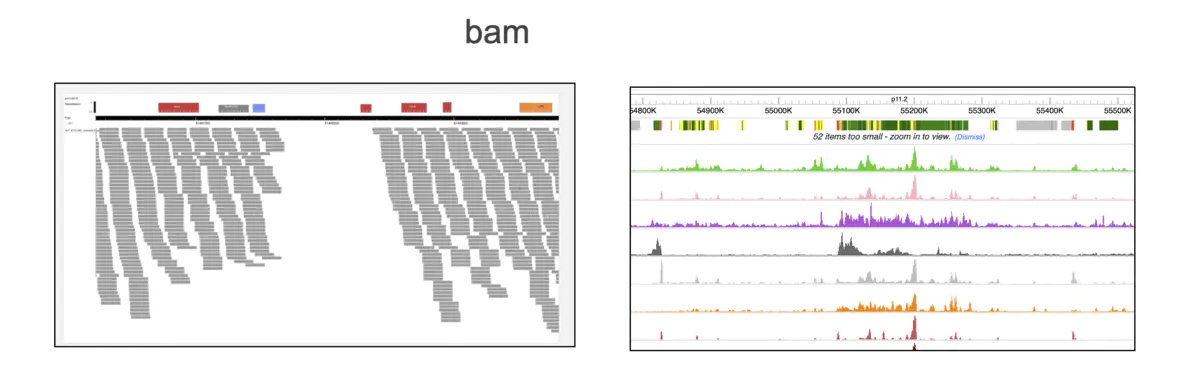
bam文件有两种显示方式,一方面是显示每条reads 的信息,另一方面是显示峰图。
bam文件必须先sort再导入washU。
甲基化数据也可以用washU来展示(如下图所示)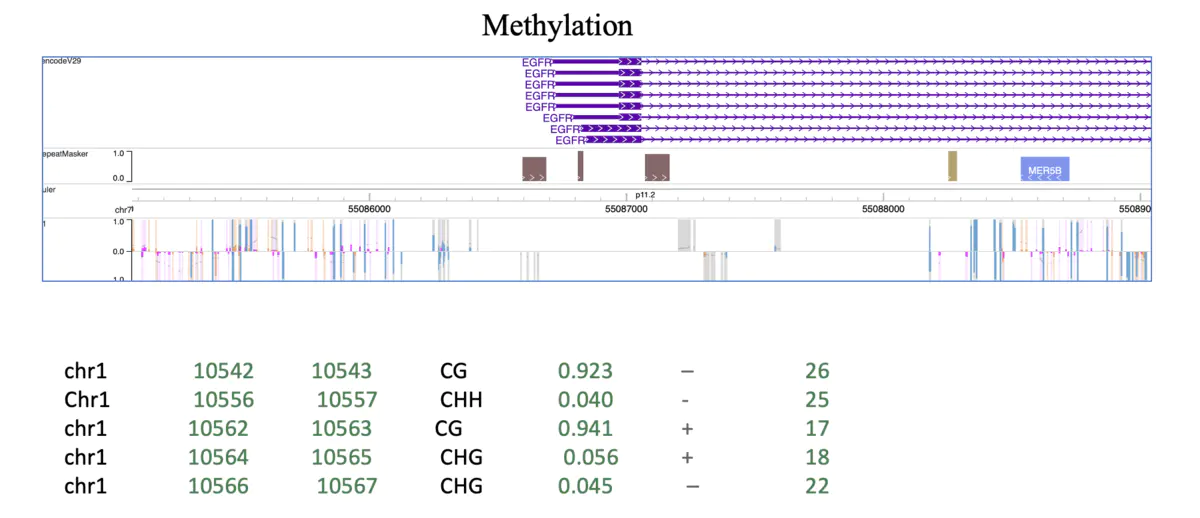
甲基化数据格式如下: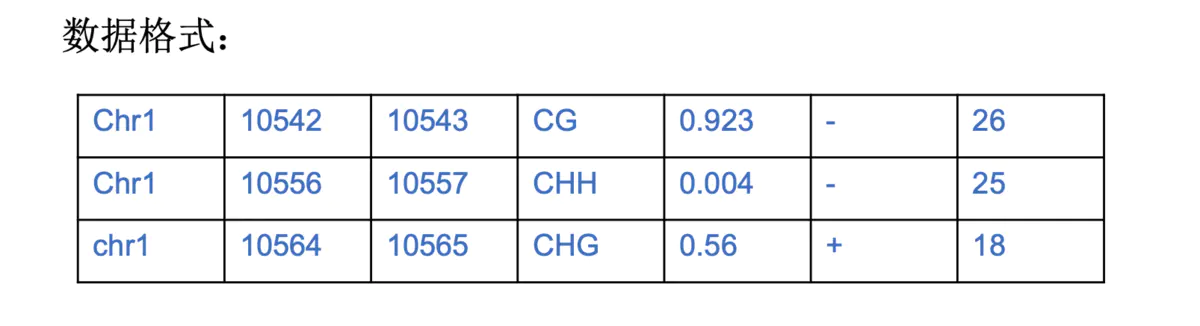 第一到三列代表甲基化位点的position
第一到三列代表甲基化位点的position
第四列代表甲基化的类型
第五列代表甲基化水平
第六列代表 正负链
第七列代表位点的覆盖深度
Hi-C数据可视化主要分为两种,一种是loop结构,一种是三角热图。其中三角热图数据采用.hic 文件格式。
.hic 文件主要是Juicer 产生的文件格式。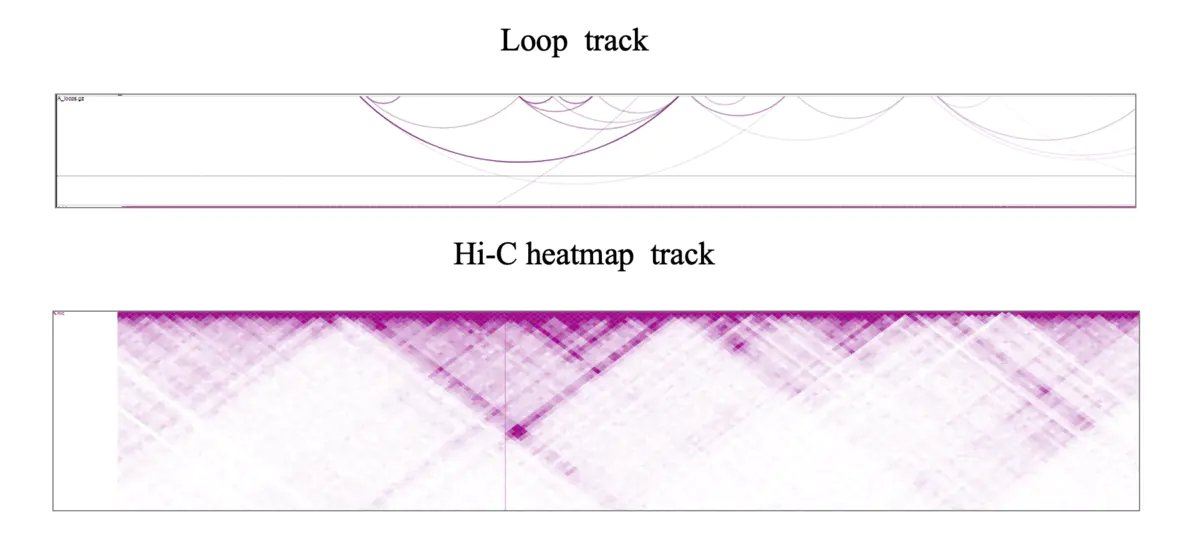
longrange
The longrange track is a bed format-like file type. Each row contains columns from left to right: chromosome, start position (0-based), and end position (not included), interaction target in this format chr2:333-444,55. As an example, interval “chr1:111-222” interacts with interval “chr2:333-444” on a score of 55, we will use following two lines to represent this interaction:
第一列: loop 左端的染色体编号
第二列:loop左端的起始位置
第三列:loop左端的终止位置
第四列:loop右端的位置以及交互数值。
此外,washU还支持calling card track 格式如下: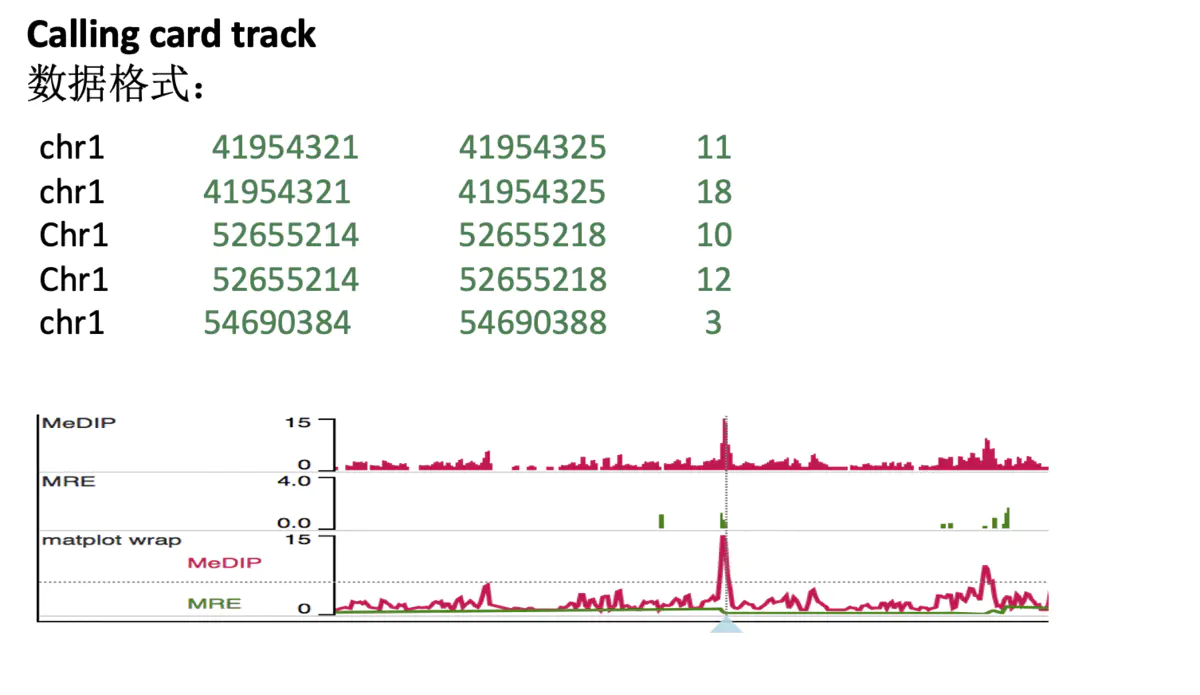 那么针对以上文件总结而言:
那么针对以上文件总结而言: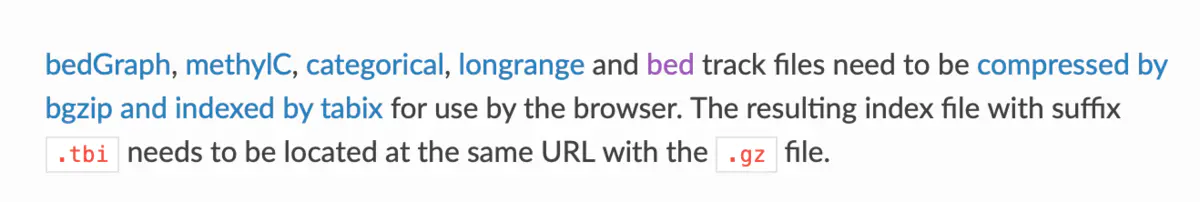
bgzip track.bedgraph.sorted
tabix -p bed track.bedgraph.sorted.gz
针对bedgraph文件都要用bgzip压缩并建立索引。
那么最后举个例子,在导入longrange,以及bam或者bigwig,我们基本就有了下面这张类似与文献里的图。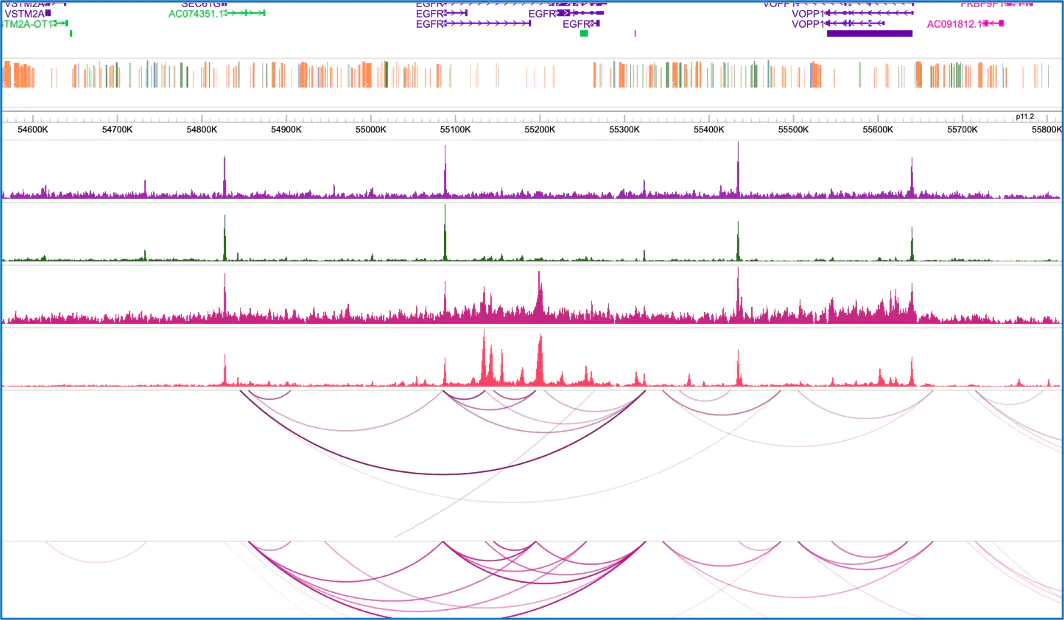
作者:XuningFan
链接:https://www.jianshu.com/p/97452f24b7cf
来源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。

若有收获,就点个赞吧
0 人点赞
