ATAC-Seq分析教程系列
ATAC-Seq分析教程:ATAC-seq的背景介绍以及与ChIP-Seq的异同
ATAC-Seq分析教程:原始数据的质控、比对和过滤
ATAC-Seq分析教程:用MACS2软件call peaks
ATAC-Seq分析教程:对ATAC-Seq/ChIP-seq的质量评估(一)phantompeakqualtools
ATAC-Seq分析教程:对ATAC-Seq/ChIP-seq的质量评估(二)ChIPQC
ATAC-Seq分析教程:重复样本的处理-IDR
ATAC-Seq分析教程:用ChIPseeker对peaks进行注释和可视化
ATAC-Seq分析教程:用网页版工具做功能分析和motif分析
ATAC-Seq分析教程:差异peaks分析——DiffBind
ATAC-Seq分析教程:ATAC-Seq、ChIP-Seq、RNA-Seq整合分析
学习目标
- 功能富集分析: [GREAT](http://bejerano.stanford.edu/great/public/html/index.php)
- motif分析:MEME套件,如DREME(http://meme-suite.org/tools/dreme)), MEME-ChIP (http://meme-suite.org/tools/meme-chip)
准备文件
提取IDR结果文件的前3列
cut -f 1,2,3 Nanog-idr-merged.bed > Nanog-idr-merged-great.bed
使用bedtools getfasta提取peaks的序列
bedtools getfasta -firef_genome.fa-bed Nanog-idr-merged-great.bed-fo Nanog-idr-merged-dreme.fasta
富集分析
GREAT(http://bejerano.stanford.edu/great/public/html/index.php)对peaks的功能注释是对peaks临近基因的注释。%E5%AF%B9peaks%E7%9A%84%E5%8A%9F%E8%83%BD%E6%B3%A8%E9%87%8A%E6%98%AF%E5%AF%B9peaks%E4%B8%B4%E8%BF%91%E5%9F%BA%E5%9B%A0%E7%9A%84%E6%B3%A8%E9%87%8A%E3%80%82)
方法:
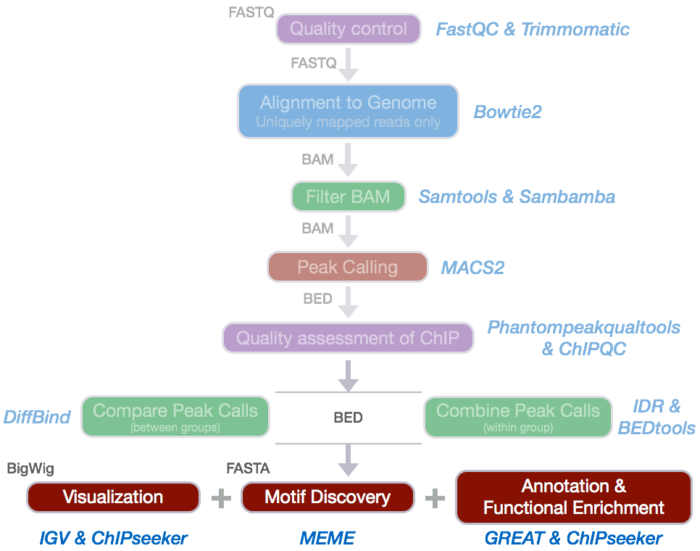
- 1.打开GREAT网站:http://bejerano.stanford.edu/great/public/html/index.php 。上传Nanog-idr-merged-great.bed文件,选择参考基因组,选择Whole genome作为背景区域,然后点击Submit。
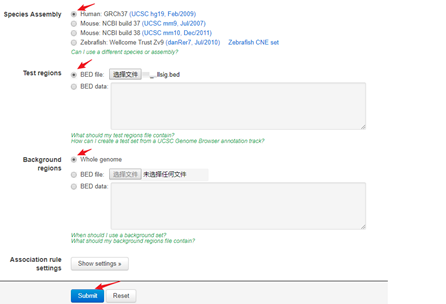
- 点开Job Description,选择View all genomic region-gene associations,结果中的两个表给出peaks注释到的基因,一个是基因组区域和基因关联的表,一个是基因和基因组关联的表:
- Region-Gene Association Graphs
这栏内容对结合位点的基因数和转录起始位点相关的基因做了图形展示。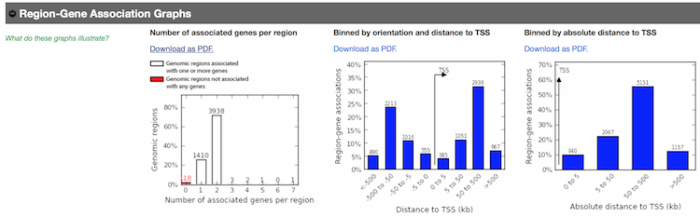
- Global Controls
选择想注释的信息,如GO注释
- 探究Nanog结合位点相关的GO BP term
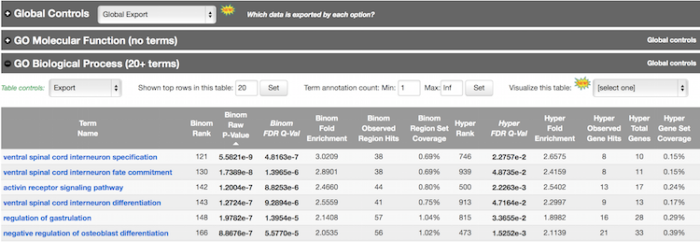
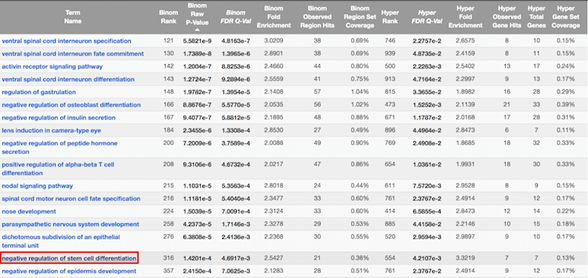
- 选择某个term,查看具体信息
- 打开This term’s genomic region-gene association tables (140 regions, 116 genes),查看注释到的GO term相关的基因,可以下载表格。
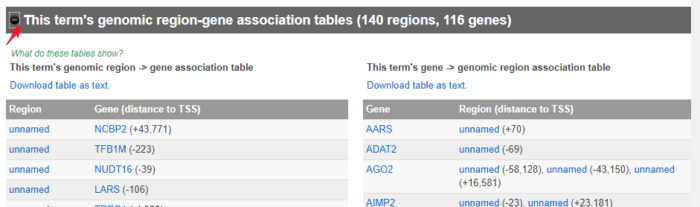
- 在This term’s gene -> genomic region association table中点击基因相关的regions,可以在UCSC浏览器上直接查看结合区域。
- 点开Job Description,选择View all genomic region-gene associations,结果中的两个表给出peaks注释到的基因,一个是基因组区域和基因关联的表,一个是基因和基因组关联的表:
找Motif
motif是比较有特征的短序列,会多次出现的,并被假设拥有生物学功能。而且,经常是一些具有序列特异性的蛋白的结合位点(如转录因子)或者是涉及到重要生物过程的(如,RNA 起始,RNA 终止, RNA 剪切等等)。
有很多网页版工具提供了找motif的方法,2014年的一篇综述列出了目前常用的网页工具(https://doi.org/10.1186/1745-6150-9-4 ):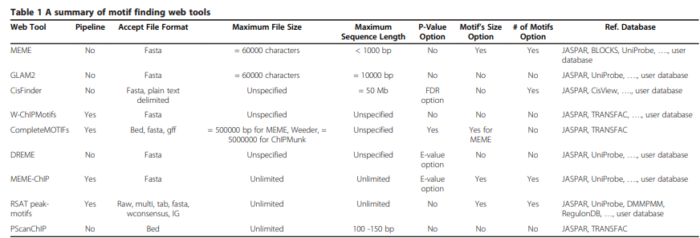
最常用的有MEME工具套件,下面主要介绍DREME。DREME适用于大批量的ChIP-Seq数据找真核转录因子短的(4nt或8nt)核心DNA结合基序。
DREME
打开DREME网页 http://meme-suite.org/tools/dreme ,只要输入fasta序列Nanog-idr-merged-dreme.fasta即可,同时可以写上email和每个任务的描述,任务完成时如右图所示,可以打开DREAM_HTML_output查看结果。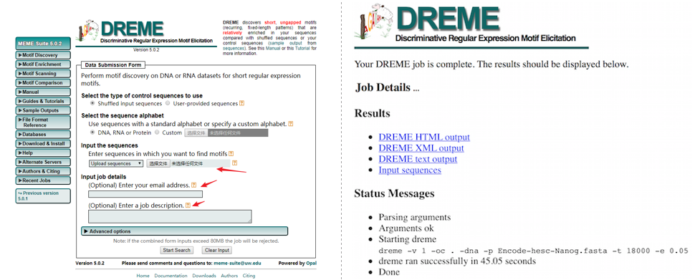
DREME’s HTML展示的结果包括找到的motif的序列logo和表示显著性的E-value。点击More可以查看更多的信息。
Tomtom
为了确定鉴定到的motif与已知转录因子的motif是否相似,可以将找到的motif再提交到Tomtom,与已知的转录因子数据库搜索匹配,同时还会给出motif-motif相似性的统计评估。
在DREAM’s HTML结果中选择某个预测到的motif,点击Submit / Download,然后选择Tomtom,点击Submit,在新打开的Tomtom界面可以选择转录因子的参考数据库,保持默认参数不变,也可以再添加其他参数,输入邮箱和任务描述就可以开始搜索。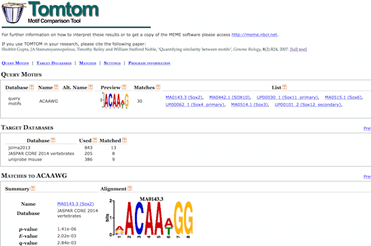
MEME-ChIP
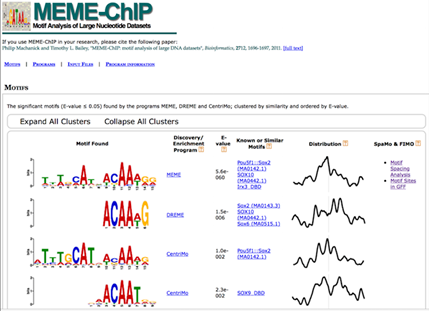
MEME-ChIP是MEME套件中的另一个工具,可以实现DREAM和Tomtom的分析功能,还可以评估motifs的中心富集性并整合相关的motifs合成相似性簇。MEME-ChIP能够鉴定更长的motifs(<30bp),但是运行时间比较长。
另外可以用Homer找motif。- 转载请务必保留本文链接:https://www.plob.org/article/24686.html

若有收获,就点个赞吧
0 人点赞
