写在前面
- 以下内容均来自我在菲沙基因(Frasergen)暑期生信培训班上记录的课堂笔记
1.Compartment计算
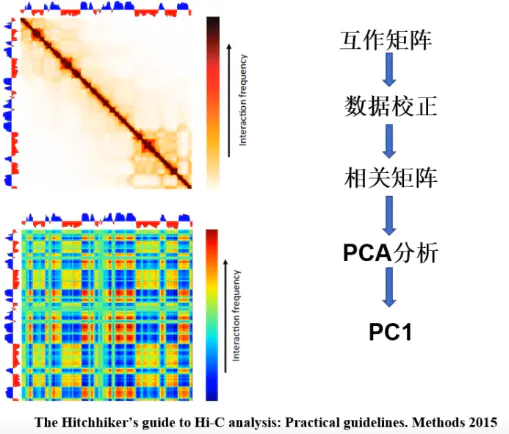
2.Compartment 分析流程
2.1 Cworld-dekker软件的安装
```bash git clone https://github.com/blajoie/cworld-dekker.gitChange directory to the
perl Build.PL ./Build ./Build install —install_base /your/custom/dir (ensure /your/custom/dir is added to your PERL5LIB path)cworld-dekkerand install thePerlmodule:
e.g.
./Build install —install_base ~/perl5
then in .bashrc
export PERL5LIB=${PERL5LIB}:/home/
<a name="nsUeL"></a>##### 2.2 分析所用数据互作图谱分染色体matrix数据,如何获得请看:[生信 | 三维基因组技术(三):Hi-C 数据比对及HiC-Pro的使用](https://www.jianshu.com/p/bb5512dfa29c)<br />matrix数据<br />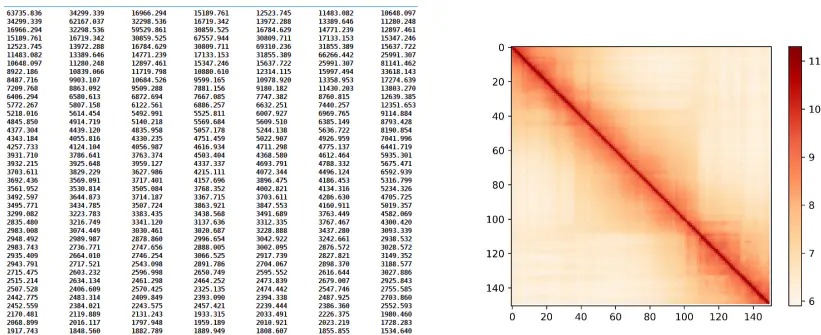<a name="z3rJ7"></a>##### 2.3 为矩阵添加headerheader文件需要自己准备,操作采用cworld的addMatrixHeaders为矩阵文件添加header```bashperl -I /software/cworld-dekker/ \/software/cworld-dekker/scripts/perl/addMatrixHeaders.pl \-i data/example.matrix \--xhf data/headerxchr1 \--yhf data/headerychr1
-I:添加cworld的库,连接到cworld软件所在目录即可
-i:matrix 文件
—xhf:横坐标表头
—yhf:纵坐标表头
自制Header文件,横纵坐标可以相同,形如: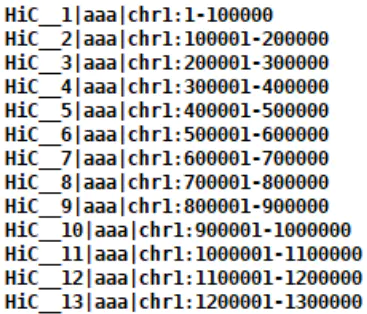
结果文件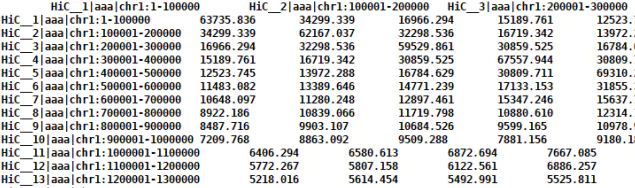
2.4 扣除背景/计算z-scale
操作采用cworld的matrix2loess.pl
#export PATH=/software/R/R-3.5.0/bin/:$PATH#export PATH=/software/bedtools/bedtools2-2.28.0/bin/:$PATHperl -I /software/cworld-dekker/ \/software/cworld-dekker/scripts/perl/matrix2loess.pl \-i example.addedHeaders.matrix.gz
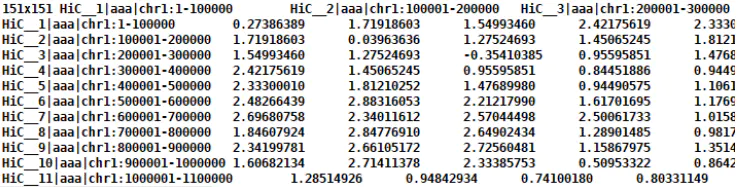
2.5 转换相关矩阵与PCA分析同时进行
采用cworld的matrix2EigenVectors.py,需要给一个example_gene.bed文件。
example_gene.bed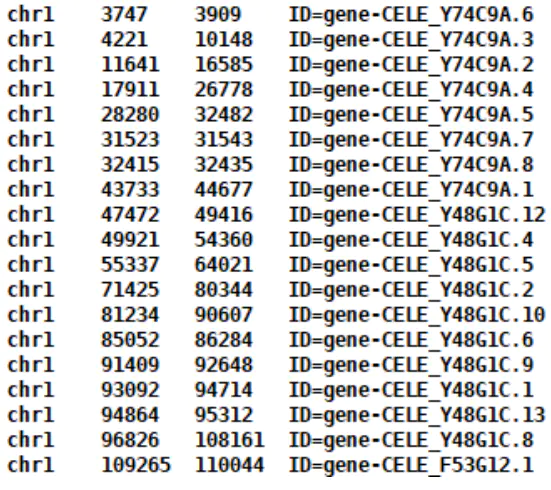
python software/cworld-dekker/scripts/python/matrix2EigenVectors.py \-i example.addedHeaders.zScore.matrix.gz \-r data/example_gene.bed
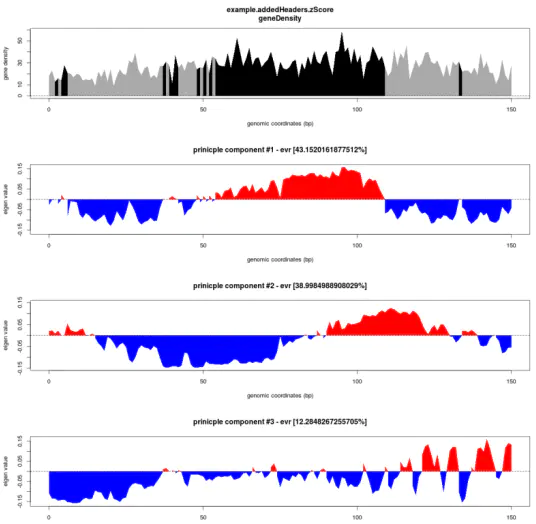
结果文件以compartments结尾。
另外生成的图片以compartments.png结尾
定义基因密度较高的区域为compartmentsA,反之为compartmentsB
作者:Bioinfo鱼
链接:https://www.jianshu.com/p/bb5512dfa29c
来源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。

若有收获,就点个赞吧
0 人点赞
