Hi-C 数据分析(番外篇一)
HiC主要写三篇相关文章:
(一)HiC的起源与发展
(二)HiC的重要应用
(三)HiC的展望
上一篇已经讲过HiC的起源与发展,这期主要讲Hi-C的实验以及数据分析。
这期主要讲Hi-C数据分析(protocol)
1. 数据过滤(质量筛选,NGS必做^_^)
2.分析HiC数据,得到自己设定分辨率(例如100kb)的热图(由于这部分需要下载基因组,网速较慢,下期介绍)
3.Hi-C数据可视化以及Find Topologically Associating Domains
(1)需要数据:Hi-C交互矩阵:N * N 的矩阵
由第二步得到的Hi-C数据是这样的格式(中间以tab分割)
| #tab chrT_001 chrT_002 chrT_003 chrT_004 chrT_001 629 164 88 105 chrT_002 164 612 175 110 chrT_003 88 175 437 105 chrT_004 105 110 105 278 |
|---|
(2)软件包:tadbit
https://github.com/zilhua/tadbit
官方网站:http://3dgenomes.github.io/TADbit/
(3)计算代码:
View Code PYTHON
| #!/user/bin/python
# -- coding:UTF-8 --
‘’’ Created on ${date} @author: www.zilhua.com ‘’’
from pytadbit import Chromosome
# initiate a chromosome object that will store all Hi-C data and analysis
#设置环境,初始化
_my_chrom = Chromosome(name=’chr19’, centromere_search=True, species=’Homo sapiens’, assembly=’NCBI36’)
###mychrom
‘’’ Chromosome chr19: 0 experiment loaded: 0 alignment loaded: species : Homo sapiens assembly version: NCBI36 ‘’’
#加载数据以BingRen的数据为例
# load Hi-C data
mychrom.add_experiment(‘k562’, cell_type=’wild type’, exp_type=’Hi-C’,
identifier=’k562’, project=’TADbit tutorial’,
hic_data=”scripts/sample_data/HIC_k562_chr19_chr19_100000_obs.txt”
, resolution=100000)
my_chrom.add_experiment(‘gm06690’,cell_type=’cancer’, exp_type=’Hi-C’,
identifier=’gm06690’, project=’TADbit tutorial’,
hic_data=”scripts/sample_data/HIC_gm06690_chr19_chr19_100000_obs.txt”, resolution=100000)
#参数解释:k562 样本名;
celltype:细胞类型#exp_type:实验类型;
hic_data:HiC数据;#resolution:分辨率
_
#对两个样本分别可视化
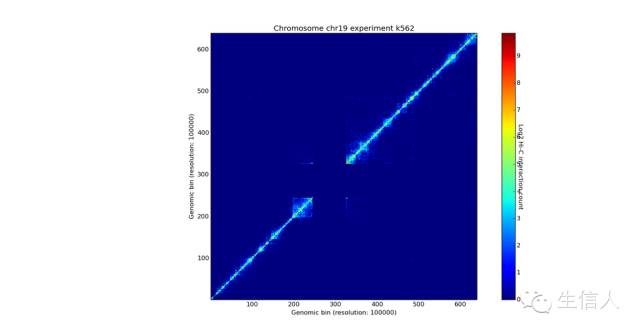
_my_chrom.experiments[“k562”].view()
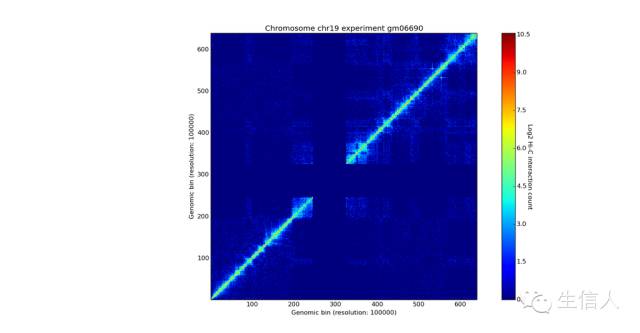
my_chrom.experiments[“gm06690”].view() |
| —- |
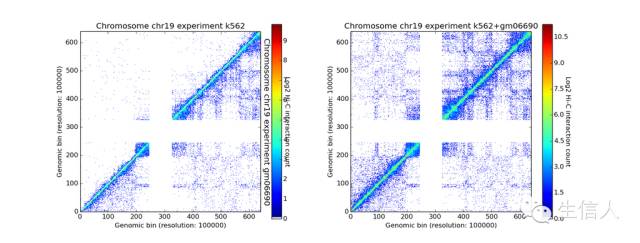
| _#如果这两个样本是对照实验,则可以这样exp = my_chrom.experiments[“k562”] + my_chrom.experiments[“gm06690”] _my_chrom.add_experiment(exp) my_chrom.visualize([(‘k562’, ‘gm06690’), ‘k562+gm06690’]) |
|---|
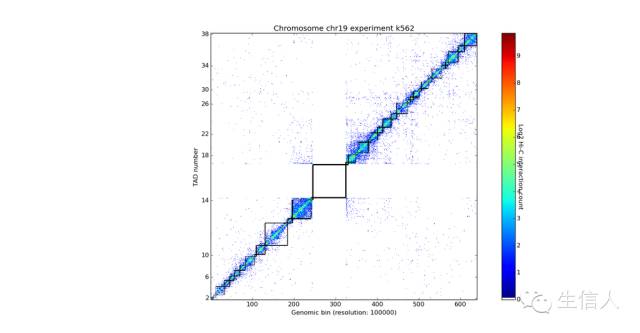
| #Find Topologically Associating Domains my_chrom.find_tad(‘k562’, n_cpus=4) _my_chrom.find_tad(‘gm06690’, n_cpus=4) #参数解释: 函数findtad第一个参数k652样本名; n_cpus线程数,个人电脑一般是1—4,服务器看配置#获取计算结果 my_chrom.experiments[“k562”].tads my_chrom.experiments[“gm06690”].tads #将数组转换成txt文本 my_chrom.experiments[“k562”].write_tad_borders() #结果中,第一列就是TAD的编号, #第二列第三列分别是TAD的起始和终止位置#第四列打分 #将TAD的边界在热图上显示 _my_chrom.visualize(‘k562’, paint_tads=True) |
|---|
| #如果需要对比两个样本的TAD情况 my_chrom.visualize([(‘k562’, ‘gm06690’)], _paint_tads=True, focus=(490,620), normalized=True) #参数解释:painttads是否描述TAD边界; #focus 显示染色体位置;normalized是否标准化 |
|---|
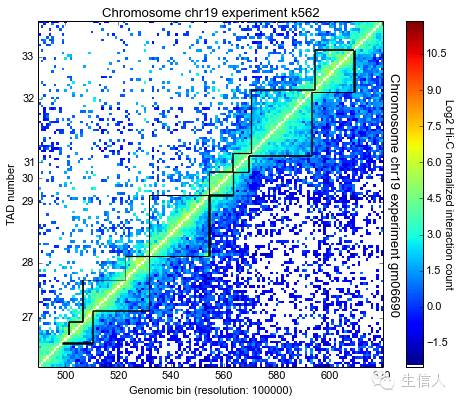
| #TAD density my_chrom.tad_density_plot(‘k562’) |
|---|

| #保存和重载数据
my_chrom.save_chromosome(“some_path.tdb”, force=True)
from pytadbit import load_chromosome
my_chrom = load_chromosome(“some_path.tdb”)
print my_chrom.experiments |
| —- |

若有收获,就点个赞吧
0 人点赞
