Hi-C简介(外传)
(一)HiC的起源与发展
(二)HiC的重要应用
(三)HiC的展望
上一篇已经讲过HiC的起源与发展,这期主要讲Hi-C的实验以及数据分析。
一、Hi-C实验
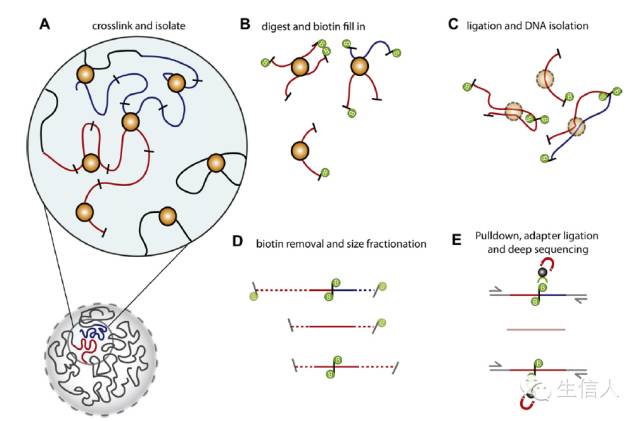
第一步:甲醛交联,固定细胞核内染色质构象
第二步:酶切,并用生物素标记
第三步:成环
第四步:打断
第五步:钓取带有生物素标记的酶切片段
二、数据分析
- 数据过滤,筛选(二代测序通用 ^__^ )
2. 比对;比对的方式主要分两种,一种判断每条reads是否含有酶切位点,有则去掉酶切位点之后的数据在进行bowtie2单端比对;另一种采用单端比对的策略,以25bp为起始长度,每次增加5bp直到该reads比对到基因组具有唯一性。
3. 寻找酶切片段;比对寻找到reads pairs在基因组物理位置之后,通过插入片段大小的限制搜索reads pairs两端每条read所对应的最近的酶切片段。酶切片段的位置代表了DNA交互产生的大致位置。
4. 筛选fragment pairs;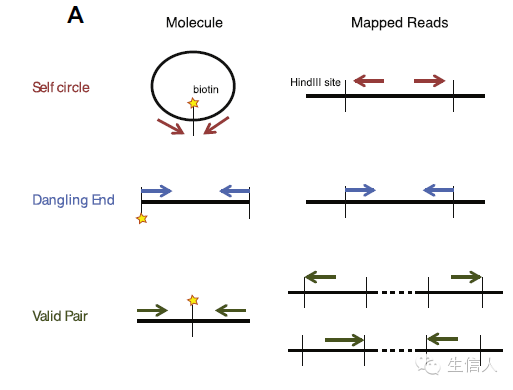
我们只需要Valid Pairs。
5.Binning;将Valid Pairs的交互信息mapping到 基因组的位置,最终转换成为每两个bin的交互强度。
6. 交互矩阵标准化;标准化方法主要分为两类,一类是基于矩阵,进行数学上的标准化,例如迭代等,另一类是基于生物学意义(例如mappingability)上的标准化。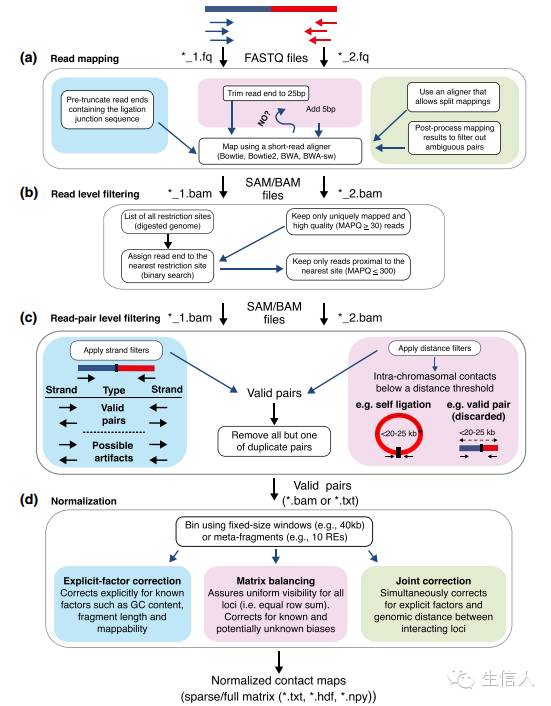
三、分析代码
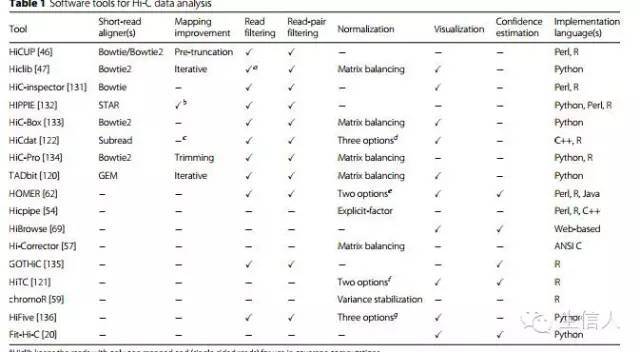
分析代码会在写完Hi-C系列所有文章之后在博客(www.zilhua.com)以protocol的形式发表,敬请关注。四、Hi-C分析工具包
欢迎关注生信人

若有收获,就点个赞吧
0 人点赞
